WO2017176265A1 - Carrier-binding agent compositions and methods of making and using the same - Google Patents
Carrier-binding agent compositions and methods of making and using the same Download PDFInfo
- Publication number
- WO2017176265A1 WO2017176265A1 PCT/US2016/026270 US2016026270W WO2017176265A1 WO 2017176265 A1 WO2017176265 A1 WO 2017176265A1 US 2016026270 W US2016026270 W US 2016026270W WO 2017176265 A1 WO2017176265 A1 WO 2017176265A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- nanoparticles
- nanoparticle
- nanoparticle composition
- lyophilized
- binding
- Prior art date
Links
Classifications
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/48—Preparations in capsules, e.g. of gelatin, of chocolate
- A61K9/50—Microcapsules having a gas, liquid or semi-solid filling; Solid microparticles or pellets surrounded by a distinct coating layer, e.g. coated microspheres, coated drug crystals
- A61K9/51—Nanocapsules; Nanoparticles
- A61K9/5107—Excipients; Inactive ingredients
- A61K9/513—Organic macromolecular compounds; Dendrimers
- A61K9/5169—Proteins, e.g. albumin, gelatin
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/30—Macromolecular organic or inorganic compounds, e.g. inorganic polyphosphates
- A61K47/42—Proteins; Polypeptides; Degradation products thereof; Derivatives thereof, e.g. albumin, gelatin or zein
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/50—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates
- A61K47/51—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent
- A61K47/62—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being a protein, peptide or polyamino acid
- A61K47/64—Drug-peptide, drug-protein or drug-polyamino acid conjugates, i.e. the modifying agent being a peptide, protein or polyamino acid which is covalently bonded or complexed to a therapeutically active agent
- A61K47/643—Albumins, e.g. HSA, BSA, ovalbumin or a Keyhole Limpet Hemocyanin [KHL]
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/50—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates
- A61K47/51—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent
- A61K47/68—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an antibody, an immunoglobulin or a fragment thereof, e.g. an Fc-fragment
- A61K47/6835—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an antibody, an immunoglobulin or a fragment thereof, e.g. an Fc-fragment the modifying agent being an antibody or an immunoglobulin bearing at least one antigen-binding site
- A61K47/6851—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an antibody, an immunoglobulin or a fragment thereof, e.g. an Fc-fragment the modifying agent being an antibody or an immunoglobulin bearing at least one antigen-binding site the antibody targeting a determinant of a tumour cell
- A61K47/6865—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an antibody, an immunoglobulin or a fragment thereof, e.g. an Fc-fragment the modifying agent being an antibody or an immunoglobulin bearing at least one antigen-binding site the antibody targeting a determinant of a tumour cell the tumour determinant being from skin, nerves or brain cancer cell
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/50—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates
- A61K47/69—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the conjugate being characterised by physical or galenical forms, e.g. emulsion, particle, inclusion complex, stent or kit
- A61K47/6921—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the conjugate being characterised by physical or galenical forms, e.g. emulsion, particle, inclusion complex, stent or kit the form being a particulate, a powder, an adsorbate, a bead or a sphere
- A61K47/6927—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the conjugate being characterised by physical or galenical forms, e.g. emulsion, particle, inclusion complex, stent or kit the form being a particulate, a powder, an adsorbate, a bead or a sphere the form being a solid microparticle having no hollow or gas-filled cores
- A61K47/6929—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the conjugate being characterised by physical or galenical forms, e.g. emulsion, particle, inclusion complex, stent or kit the form being a particulate, a powder, an adsorbate, a bead or a sphere the form being a solid microparticle having no hollow or gas-filled cores the form being a nanoparticle, e.g. an immuno-nanoparticle
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/0012—Galenical forms characterised by the site of application
- A61K9/0019—Injectable compositions; Intramuscular, intravenous, arterial, subcutaneous administration; Compositions to be administered through the skin in an invasive manner
Definitions
- This application relates to novel compositions of binding agents and carrier proteins and methods of making and using the same, in particular, as a cancer therapeutic.
- Chemotherapy remains a mainstay for systemic therapy for many types of cancer, including melanoma. Most chemotherapeutics are only slightly selective to tumor cells, and toxicity to healthy proliferating cells can be high (Allen TM. (2002) Cancer 2:750-763), often requiring dose reduction and even discontinuation of treatment.
- one way to overcome chemotherapy toxicity issues as well as improve drug efficacy is to target the chemotherapy drug to the tumor using antibodies that are specific for proteins selectively expressed (or overexpressed) by tumors cells to attract targeted drugs to the tumor, thereby altering the biodistribution of the chemotherapy and resulting in more drug going to the tumor and less affecting healthy tissue.
- specific targeting rarely succeeds in the therapeutic context.
- ADC antibody dependent chemotherapy
- Antibody-targeted chemotherapy promised advantages over conventional therapy because it provides combinations of targeting ability, multiple cytotoxic agents, and improved therapeutic capacity with potentially less toxicity.
- clinically effective antibody -targeted chemotherapy remains elusive: major hurdles include the instability of the linkers between the antibody and chemotherapy drug, reduced tumor toxicity of the chemotherapeutic agent when bound to the antibody, and the inability of the conjugate to bind and enter tumor cells.
- these therapies did not allow for control over the size of the antibody-drug conjugates.
- composition in addition, as to any therapeutic application, there also remains a need for the composition to be stable in its physical, chemical and biological properties.
- Lyophilization removes water from a composition.
- the material to be dried is first frozen and then the ice or frozen solvent is removed by sublimation in a vacuum environment.
- An excipient may be included in pre-lyophilized formulations to enhance stability during the freeze-drying process and/or to improve stability of the lyophilized product upon storage. Pikal, M. Biopharm. 3(9)26-30 (1990) and Arakawa et al., Pharm. Res. 8(3):285- 291 (1991).
- proteins may be lyophilized, the process of lyophilization and reconstitution may affect the properties of the protein. Because proteins are larger and more complex than traditional organic and inorganic drugs (i.e. possessing multiple functional groups in addition to complex three-dimensional structures), the formulation of such proteins poses special problems. For a protein to remain biologically active, a formulation must preserve intact the conformational integrity of at least a core sequence of the protein's amino acids while at the same time protecting the protein's multiple functional groups from degradation. Degradation pathways for proteins can involve chemical instability (i.e. any process which involves modification of the protein by bond formation or cleavage resulting in a new chemical entity) or physical instability (i.e. changes in the higher order structure of the protein).
- chemical instability i.e. any process which involves modification of the protein by bond formation or cleavage resulting in a new chemical entity
- physical instability i.e. changes in the higher order structure of the protein.
- Chemical instability can result from deamidation, racemization, hydrolysis, oxidation, beta elimination or disulfide exchange. Physical instability can result from denaturation. aggregation, precipitation or adsorption, for example. The three most common protein degradation pathways are protein aggregation, deamidation and oxidation. Cleland, et al., Critical Reviews in Therapeutic Drug Carrier Systems 10(4): 307-377 (1993). SUMMARY
- the composition comprises nanoparticles which contain (a) carrier protein (b) a binding agent, and (c) optionally a therapeutic agent.
- the binding agent is believed to be bound to the carrier protein through hydrophobic interactions which, by their nature, are weak. Yet the activity of the individual components, as well as their relative relationship in the nanoparticle are preserved despite lyophilization and reconstitution of the a composition.
- binding to the carrier protein occurs through some or all of the hydrophobic portion of the binding agent, e.g., the Fc component, which results in all or part of the hydrophobic portion being integrated into the carrier protein core, while the target binding portions (regions) (e.g., an Fa and Fb portion) of the antibody remain outside of the carrier protein core, thereby retaining their target specific binding capabilities.
- the binding agent is a non-therapeutic and non-endogenous human antibody, a fusion protein, e.g., fusion of an antibody Fc domain to a peptide that binds a target antigen, or an aptamer.
- the size of nanoparticles, and the distribution of the size, is also important.
- Nanoparticles may behave differently according to their size. At large sizes, nanoparticles or the agglomeration of the particles may block blood vessels either of which can affect the performance and safety of the composition.
- the inventive compositions comprise nanoparticles which nanoparticles contain (a) carrier protein (b) a binding agent and (c) optionally a therapeutic agent.
- the binding agent is believed to be bound to the carrier protein through hydrophobic interactions which, by their nature, are weak. Yet, the activity of the individual components, and their relative relationship in the nanoparticle are still achieved despite lyophilization and reconstitution of the composition.
- nanoparticle compositions comprising
- each of the nanoparticles comprises a carrier protein, between about 100 to about 1000 binding agents, and optionally at least one therapeutic agent, wherein the binding agents are arranged outward from the surface of the nanoparticles and wherein the nanoparticles are capable of binding to a predetermined epitope in vivo.
- large particles e.g. greater than 1 ⁇
- larger particles can accumulate in the tumor or specific organs. See e.g. 20-60 micron glass particle that is used to inject into the hepatic artery feeding a tumor of the liver, called "TheraSphere" (in clinical use for liver cancer).
- particles under 1 ⁇ are used for intravenous administration. Particles over 1 ⁇ are, more typically, administered directly into a tumor ("direct injection") or into an artery feeding into the site of the tumor.
- nanoparticle compositions comprising nanoparticles wherein each of the nanoparticles comprises a carrier protein that is not albumin, between about 100 to about 1000 binding agents, preferably about 400 to about 800 binding agents, and optionally at least one therapeutic agent, wherein the binding agents are arranged on an outside surface of the nanoparticles and wherein the nanoparticles are capable of binding to a predetermined epitope in vivo.
- the number of binding agents is increased proportionally. For example, if a 160 nm nanoparticle contains 400 binding agents, a 320 nm dimer contains about 800 binding agents.
- nanoparticle compositions comprising nanoparticles, wherein each of the nanoparticles comprises carrier protein, between about 400 to about 800 binding agents, and optionally at least one therapeutic agent that is not paclitaxel, wherein the binding agents are arranged on a surface of the nanoparticles such that the binding portion of the binding agent is directed outward from that surface and wherein the nanoparticles are capable of binding to a predetermined epitope in vivo.
- the nanoparticles multimerize, e.g. dimerize. Multimerization may be observed as multiples of the weight or size of the unit molecule, e.g. 160 nm particles multimerize to about 320 nm, 480 nm, 640 nm, etc. In some embodiments, less than 20% of the nanoparticles in a population are multimers. In some embodiments, more than 80% of the nanoparticles in a population are multimers.
- the weight ratio of carrier-bound drug to binding agent is between about 5: 1 to about 1 : 1. In one embodiment, the weight ratio of carrier-bound drug to binding agent is about 10:4.
- the binding agents are a substantially single layer on all or part of the surface of the nanoparticle. In one embodiment, less than 0.01% of nanoparticles in the composition have a size selected from the group consisting of greater than 200 nm, greater than 300 nm, greater than 400 nm, greater than 500 nm, greater than 600 nm, greater than 700 nm and greater than 800 nm. Larger sizes are believed to be the result of multimerization of several nanoparticles, each comprising a core and binding agent coating on all or part of the surface of each nanoparticle.
- the invention further includes lyophilized compositions, and lyophilized
- compositions that do not materially differ from, or are the same as, the properties of freshly- prepared nanoparticles.
- the lypholized composition upon resuspending in aqueous solution, is similar or identical to the fresh composition in terms of particle size, particle size distribution, toxicity for cancer cells, binding agent affinity, and binding agent specificity.
- the invention is directed to the surprising finding that lyophilized nanoparticles retain the properties of freshly-made nanoparticles after resuspension notwithstanding the presence of two different protein components in these particles.
- this invention relates to a lyophilized nanoparticle composition
- a lyophilized nanoparticle composition comprising nanoparticles, wherein each of the nanoparticles comprises a carrier-bound drug core and an amount of binding agent arranged on a surface of the core such that the binding portion of the binding agent is directed outward from that surface, wherein the binding agents retain their association with the outside surface of the nanoparticle upon reconstitution with an aqueous solution.
- the lyophilized composition is stable at room temperature for at least 3 months.
- the reconstituted nanoparticles retain the activity of the therapeutic agent and are capable of binding to the target in vivo.
- the average reconstituted nanoparticle size is from about 130 nm to about 1 ⁇ . In a preferred embodiment, the average reconstituted nanoparticle size is from about 130 nm to about 200 nm, and more preferably about 160 nm. In one embodiment, in the average reconstituted nanoparticle size is from greater than 800 nm to about 3.5 ⁇ , comprising multimers of smaller nanoparticles, e.g. multimers of 100-200 nm nanoparticles. In one embodiment, the weight ratio of core to binding agent is from greater than 1 : 1 to about 1 :3. In one embodiment, in the average reconstituted nanoparticle size is about 160nm to about 225nm.
- this disclosure relates to a lyophilized nanoparticle composition
- a lyophilized nanoparticle composition comprising nanoparticles, wherein each of the nanoparticles comprises: (a) an albumin-bound paclitaxel core and (b) between about 400 to about 800 molecules of bevacizumab arranged on a surface of the albumin-bound paclitaxel core such that the binding portion of the binding agent is directed outward from that surface, wherein the binding agents retain their association with the surface of the nanoparticle upon reconstitution with an aqueous solution, provided that said lyophilized composition is stable at about 20 °C to about 25 °C for at least 3 months and the reconstituted nanoparticles are capable of binding to VEGF in vivo.
- this disclosure relates to a lyophilized nanoparticle composition
- a lyophilized nanoparticle composition comprising nanoparticles, wherein each of the nanoparticles comprises: (a) an albumin-bound paclitaxel core and (b) an amount of bevacizumab arranged on a surface of the albumin- bound paclitaxel core such that the binding portion of the binding agent is directed outward from that surface, wherein the binding agents retain their association with the surface of the nanoparticle upon reconstitution with an aqueous solution, provided that said lyophilized composition is stable at about 20 °C to about 25 °C for at least 3 months and the reconstituted nanoparticles are capable of binding to VEGF in vivo, and further wherein the average reconstituted nanoparticle size is not substantially different from the particle size of the freshly prepared nanoparticles.
- the average particle sizes are between 200 and 800 nm, including 200, 300, 400, 500, 600, 700 or 800nm. In other embodiments, the average particles are larger, e.g. from greater than 800 nm to about 3.5 ⁇ . In some embodiments, the particles are multimers of nanoparticles. In some embodiments the nanoparticles have average particle sizes of about 160nm to about 225 nm either freshly made or after lyophilization and resuspension in an aqueous solution suitable for injection.
- the weight ratio of albumin-bound paclitaxel to bevacizumab is between about 5: 1 to about 1 : 1. In other embodiments, the weight ratio of albumin-bound paclitaxel to bevacizumab is about 10:4. In further embodiments, the weight ratio of albumin- bound paclitaxel to bevacizumab is from greater than 1 : 1 to about 1 :3.
- the core is albumin-bound paclitaxel
- the binding agents are selected from binding agents that selectively recognize VEGF (e.g. bevacizumab/ Avastin), binding agents that selectively recognize CD20 (e.g. rituximab/Rituxin) and binding agents that selectively recognize Her2 (Trastuzumab/Herceptin).
- the at least one therapeutic agent is located inside the nanoparticle. In other embodiments, the at least one therapeutic agent is located on the outside surface of the nanoparticle. In yet other embodiments, the at least one therapeutic agent is located inside the nanoparticle and on the outside surface of the nanoparticle.
- the nanoparticle contains more than one type of therapeutic agent.
- a taxane and a platinum drug e.g. paclitaxel and cisplatin.
- the binding agents are selected from the group consisting of ado- trastuzumab emtansine, alemtuzumab, bevacizumab, cetuximab, denosumab, dinutuximab, ipilimumab, nivolumab, obinutuzumab, ofatumumab, panitumumab, pembrolizumab, pertuzumab, rituximab, and trastuzumab.
- the binding agents are a substantially single layer of binding agents on all or part of the surface of the nanoparticle.
- the antibodies are less glycosylated than normally found in natural human antibodies.
- Such glycosylation can be influenced by e.g. the expression system, or the presence of glycosylation inhibitors during expression.
- the glycosylation status of an antibody or other binding agent is altered through enzymatic or chemical action.
- the at least one therapeutic agent is selected from the group consisting of abiraterone, bendamustine, bortezomib, carboplatin, cabazitaxel, cisplatin, chlorambucil, dasatinib, docetaxel, doxorubicin, epirubicin, erlotinib, etoposide, everolimus, gefitinib, idarubicin, imatinib, hydroxyurea, imatinib, lapatinib, leuprorelin, melphalan, methotrexate, mitoxantrone, nedaplatin, nilotinib, oxaliplatin, paclitaxel, pazopanib, pemetrexed, picoplatin, romidepsin, satraplatin, sorafenib, vemurafenib, sunitinib, tenipos
- the nanoparticle further comprises at least one additional therapeutic agent that is not paclitaxel or bevacizumab.
- the binding agents, carrier protein and, when present, therapeutic agent are bound through non-covalent bonds.
- the carrier protein is selected from the group consisting of gelatin, elastin, gliadin, legumin, zein, a soy protein, a milk protein, and a whey protein.
- the carrier protein is albumin, for example, human serum albumin.
- the composition is formulated for intravenous delivery. In other embodiments, the composition is formulated for direct injection or perfusion into a tumor.
- the average nanoparticle size in the composition is from greater than 800 nm to about 3.5 ⁇ .
- the nanoparticles have a dissociation constant between about 1 x 10 "11 M and about 1 x 10 "9 M.
- nanoparticle compositions comprising contacting the carrier protein and the optionally at least one therapeutic agent with the antibodies in a solution having a pH of between 5.0 and 7.5 and a temperature between about 5°C and about 60°C, between about 23°C and about 60°C, or between about 55°C and about 60°C under conditions and ratios of components that will allow for formation of the desired nanoparticles.
- the nanoparticle is made at 55- 600C and pH 7.0.
- nanoparticle compositions comprising (a) contacting the carrier protein and optionally the at least one therapeutic agent to form a core and (b) contacting the core with the antibodies in a solution having a pH of about 5.0 to about 7.5 at a temperature between about 5°C and about 60°C, between about 23°C and about 60°C, or between about 55°C and about 60°C under conditions and ratios of components that will allow for formation of the desired nanoparticles.
- the amount of components is controlled in order to provide for formation of the desired nanoparticles.
- a composition wherein the amount of components is too dilute will not form the nanoparticles as described herein.
- weight ratio of carrier protein to binding agent is 10:4.
- the amount of carrier protein is between about 1 mg/mL and about 100 mg/mL.
- the amount of binding agent is between about 1 mg/mL and about 30 mg/mL.
- the ratio of carrier protein: binding agent: solution is approximately 9 mg of carrier protein (e.g., albumin) to 4 mg of binding agent (e.g., BEV) in 1 mL of solution (e.g., saline).
- An amount of therapeutic agent e.g., paclitaxel can also be added to the carrier protein.
- the nanoparticles are made as above, and then lyophilized.
- methods for treating a cancer cell comprising contacting the cell with an effective amount of a nanoparticle composition disclosed herein to treat the cancer cell.
- a tumor in a patient in need thereof comprising contacting the cell with an effective amount of a nanoparticle composition disclosed herein to treat the tumor.
- the size of the tumor is reduced.
- the nanoparticle composition is administered intravenously.
- the nanoparticle composition is administered by direct injection or perfusion into the tumor.
- the methods provided herein include the steps of: a) administering the nanoparticle composition once a week for three weeks; b) ceasing administration of the nanoparticle composition for one week; and c) repeating steps a) and b) as necessary to treat the tumor.
- the treatment comprises administration of the targeting binding agent prior to administration of the nanoparticles.
- the targeting binding agent is administered between about 6 and 48, or 12 and 48 hours prior to
- the targeting binding agent is administered between 6 and 12 hours prior to administration of the nanoparticles. In yet another embodiment, the targeting binding agent is administered between 2 and 8 hours prior to administration of the nanoparticles. In still other embodiments, the targeting binding agent is administered a week prior to administration of the nanoparticles. For example,
- pretreatment with BEV may comprise administration of lmg/kg BEV which is 1/lOth the usual dose, followed by administration of AB160.
- the therapeutically effective amount comprises about 75 mg/m 2 to about 175 mg/m 2 of the carrier protein (i.e., milligrams carrier protein per m 2 of the patient). In other embodiments, the therapeutically effective amount comprises about 75 mg/m 2 to about 175 mg/m 2 of therapeutic agent (e.g., paclitaxel). In other embodiments, the therapeutically effective amount comprises about 30 mg/m 2 to about 70 mg/m 2 of the binding agent. In yet other embodiments, the therapeutically effective amount comprises about 30
- the lypholized composition comprises from about 75
- the carrier protein which is preferably albumin; from about
- paclitaxel from about 75 mg/m to about 175 mg/m of paclitaxel.
- An embodiment of the invention includes a method for increasing the duration of tumor uptake of a chemotherapeutic agent by administering the chemotherapeutic agent in a nanoparticle comprising a carrier protein and the chemotherapeutic agent having surface complexation with an antibody, e.g., an antibody that specifically binds to an antigen on or shed by the tumor.
- an antibody e.g., an antibody that specifically binds to an antigen on or shed by the tumor.
- FIG. 1A shows flow cytometry scatterplots including: ABRAXANE® (ABX - commercially available from Celgene Corporation, Summit, NJ 07901) stained with secondary antibody only (top left panel), ABX stained with goat anti-mouse IgGl Fab plus secondary antibody (top right panel), AB160 (which is a nanoparticle of albumin-bound paclitaxel to bevacizumab in a ratio of about 10:4 and has an average particle size of 160 nm) stained with secondary antibody only (bottom left panel), or AB 160 stained with goat anti-mouse IgGl Fab plus secondary antibody (bottom right panel).
- ABRAXANE® ABX - commercially available from Celgene Corporation, Summit, NJ 07901
- ABX stained with goat anti-mouse IgGl Fab plus secondary antibody top right panel
- AB160 which is a nanoparticle of albumin-bound paclitaxel to bevacizumab in a ratio of about 10:4 and has
- FIG. IB shows a representative electron micrograph after incubation of AB160 with gold particle-labeled anti-human IgG Fe.
- FIG. 1C shows a pie chart (top) indicating the percentages of total paclitaxel in AB 160 fractions (particulate, proteins greater than 100 kD and proteins less than 100 kD); and a Western blot with antibodies against mouse IgG Fab (BEV) and paclitaxel to verify co-localization (bottom).
- FIG. 1 D represents the activity of paclitaxel in an in vitro toxicity assay with A375 human melanoma cells, compared to ABX alone. The results are represented by the average (+/- SEM) proliferation index, which is the percentage of total proliferation of untreated cells. This data represents 3 experiments and differences were not significant.
- FIG. IE represents results from a VEGF ELISA of supernatant after co-incubation of VEGF with ABX and AB160 to determine binding of the ligand, VEGF, by the antibody.
- the results are shown as the average percentage +/- SEM of VEGF that was unbound by the 2 complexes.
- the data represents 3 experiments ** P ⁇ 0.005.
- FIG. 2A shows the size of the complexes (determined by light scattering technology) formed by adding BEV (bevacizumab) to ABX under conditions where nanoparticles and higher are formed. Increasing concentrations of BEV (0-25 mg) were added to 10 mg of ABX and the size of the complexes formed was determined. The average size of the complexes (146 nm to 2, 166 nm) increased as the concentration of BEV was increased. The data is displayed as volume of sample/size and graphs show the size distribution of the particles. This data is representative of 5 separate drug
- ABX by itself, has an average particle size of about 130 nm.
- FIG. 2B shows affinity of the binding of ABX and BEV (as determined by light absorption (BLItz) technology).
- the data is displayed as dissociation constant (Kd).
- Kd dissociation constant
- FIG. 2C shows the stability of the nanoparticle complexes from Figure 2B in serum as determined by a nanoparticle tracking analysis (NTA) on Nanosight 300
- FIG. 3A shows in vivo testing of AB nanoparticles in athymic nude mice injected
- FIG. 3B shows Kaplan-Meier curves generated for median survival of the mice analyzed in FIG. 3 A. Significance was determined using the Mantle-Cox test comparing survival curves.
- FIG. 3C shows the percent change from baseline for mice treated when tumors
- FIG. 3D shows in vivo testing of AB nanoparticles in athymic nude mice injected
- FIG. 3E shows Kaplan-Meier curves generated for median survival of the mice analyzed in FIG. 3D. Significance was determined using the Mantle-Cox test comparing survival curves.
- FIG. 4A demonstrates blood paclitaxel concentration displayed in line graph with y-axis in log scale, based on blood and tumor samples taken from non-tumor and tumor bearing mice at 0-24 hours after IV injection with 30 mg/kg of paclitaxel in the context of ABX or AB 160 and measured by LC-MS.
- Mice were IV injected at time 0, with blood samples taken and the mice sacrificed at time points of 0, 4, 8, 12, and 24 hours. There were at least 3 mice per time point. Student's t-test was utilized to determine if any differences in concentrations between ABX and AB 160 were significant.
- FIG. 4B demonstrates the blood paclitaxel concentration from FIG. 4A, displayed in line graph with y-axis in numeric scale.
- FIG. 4C shows the C max , half-life and AUC values calculated from the blood concentration data provide in FIGs 4 A and 4B.
- FIG. 4D demonstrates blood paclitaxel concentration displayed in line graph with y-axis in log scale from a second pharmacokinetic experiment using earlier time points (2 to 8 hours).
- FIG. 4E demonstrates the blood paclitaxel concentration from FIG. 4D, displayed in line graph with y-axis in numeric scale.
- FIG. 4F shows blood paclitaxel concentration in mice in which the tumors were allowed to grow to a larger size before ABX and AB 160 injections.
- FIG. 4G shows the ⁇ note and the AUC calculated from the data in FIG. 4F.
- FIG. 4H shows paclitaxel concentrations in the tumors from the second mouse experiment as determined by LC-MS. Data are displayed as ⁇ g of paclitaxel/mg of tumor tissue. Student's t-test was utilized to determine if differences were significant.
- FIG. 41 shows 1-125 radioactivity levels in mice treated with AB160 relative to ABX alone.
- FIG. 4J shows a graphical representation of the 1-125 radioactivity levels shown in FIG. 41.
- FIG. 5A shows particle size measurements and affinity of nanoparticles made with rituximab.
- lO mg/ml of ABX was incubated with rituximab (RIT) at 0-10 mg/ml and light scatter technology (Mastersizer 2000) was used to determine resulting particle sizes.
- Data are displayed as the percent volume of particles at each size and the curves represent particle size distributions (top).
- the table (bottom) shows the sizes of the resulting particles at each concentration of antibody.
- FIG. 5B shows particle size measurements and affinity of nanoparticles made with trastuzumab. 10 mg/ml of ABX was incubated with trastuzumab (HER) at 0-22 mg/ml and light scatter technology (Mastersizer 2000) was used to determine resulting particle sizes. Data are displayed as the percent volume of particles at each size and the curves represent particle size distributions (top). The table (bottom) shows the sizes of the resulting particles at each concentration of antibody.
- FIG. 5C shows the binding affinity of rituximab and trastuzumab as compared to ABX at pH 3, 5, 7 and 9, determined by biolayer interferometry (BLitz) technology. The dissociation constants are displayed for each interaction.
- FIG. 6A shows in vitro toxicity of AR160 as tested with the CD20-positive Daudi human lymphoma cell line.
- the data are displayed in a graph of the proliferation index, which is the percent of FITC positive cells in treated wells relative to FITC positive cells in the untreated well (the highest level of proliferation).
- FIG. 6B shows in vivo tumor efficacy in athymic nude mice injected with 5 x
- mice were treated with PBS, 30 mg/kg ABX, 12 mg/kg rituximab, 12 mg/kg rituximab + 30 mg/kg ABX, or AR160.
- Tumor response was determined at day 7 post-treatment by the percent change in tumor size from the first day of treatment. Significance was determined by Student's t-test; the percent change from baseline was significantly different between the AR160 treated mice and all other groups (pO.0001).
- FIG. 6C shows Kaplan-Meier survival curves generated from the experiment shown in FIG. 6B. Median survival for each treatment group is shown. A Mantle-Cox test was used to determine whether survival curve differences were significant.
- FIG. 7A demonstrates addition of another chemotherapy drug (cisplatin) to AB160.
- ABX (5 mg/ml) was incubated with cisplatin (0.5 mg/ml) at room temperature for 30 minutes and free cisplatin was measured by HPLC in the supernatant after ABX particulate was removed. The quantity of free cisplatin was subtracted from the starting concentration to determine the quantity of cisplatin that bound to the ABX. The data are displayed in a column graph, along with the starting concentration (cisplatin).
- FIG. 7B shows the toxicity of cisplatin-bound ABX (AC) in a proliferation assay of A375 human melanoma cells. After 24 hours of drug exposure and EdU
- the cells were fixed, permeabilized and labeled with a FITC conjugated anti-EdU antibody.
- the data is displayed in a graph of the proliferation index, which is the percent of FITC positive cells in treated wells compared to FITC positive cells in the untreated well (the highest level of proliferation).
- FIG. 7C shows in vivo tumor efficacy of AC (ABC complex; cisplatin-bound
- the tumors were allowed to grow to 600 mm to 900 mm and the mice were treated with PBS, 30 mg/kg ABX, 2 mg/kg cisplatin, AB 160, 2 mg/kg cisplatin + AB160 or ABC 160.
- Tumor response was determined at day 7 post-treatment by the percent change in tumor size from the day of treatment. Significance was determined by Student's t-test; the percent change from baseline was significantly different between the ABC 160 treated mice and PBS-, cisplatin-, or ABX-treated mice (pO.0001). There was no significant difference between the AB160, AB 160 + cisplatin, and ABC160 treated groups for day 7 post-treatment percent change from baseline.
- FIG. 7D shows Kaplan-Meier survival curves generated based on the experiment shown in FIG. 7C and median survival for each treatment group is shown. A Mantle- Cox test was used to determine whether survival curve differences were significant.
- FIG. 8A shows the size distribution of AB160 nanoparticles that were lyophilized, stored at room temperature for one month, and reconstituted, as compared to fresh AB160 or ABX alone.
- FIG. 8B shows the ligand (VEGF) binding ability of AB 160 nanoparticles that were lyophilized, stored at room temperature for one month, and reconstituted, as compared to fresh AB160 or ABX alone.
- FIG. 8C shows in vitro cancer cell toxicity of AB160 nanoparticles that were lyophilized, stored at room temperature for one month, and reconstituted, as compared to fresh AB160 or ABX alone.
- FIG. 8 D shows the size distribution of AB160 nanoparticles that were
- FIG. 8 E shows the ligand (VEGF) binding ability of AB160 nanoparticles that were lyophilized, stored at room temperature for ten months, and reconstituted, as compared to fresh AB160 or ABX alone.
- FIG. 8 F shows in vitro cancer cell toxicity of AB160 nanoparticles that were lyophilized, stored at room temperature for ten months, and reconstituted, as compared to fresh AB160 or ABX alone.
- FIGs. 9A-C show the size distributions of the ABX-BEV complexes at IV. infusion conditions (ABX final concentration of 5 mg/mL) incubated in saline at room temperature for up to 24 hours (FIGs. 9A and 9B). By 4 hours at room temperature, there is some evidence of complex breakdown by ELISA (20%, FIG. 9 C).
- FIG. 10 shows in vitro incubation for 30 seconds of ABX (top panel) or AB160 (bottom panel) in saline or heparinized human plasma at relative volume ratios of 9: 1 or 1 : 1.
- FIGs. 11 A-E show in vivo testing of athymic nude mice injected with 1 x 10 A375 human melanoma cells in the right flank and treated with (FIG 11 A) PBS, (FIG 11B) 12 mg/kg BEV, (FIG 1 1 C) 30 mg/kg ABX, (FIG 1 1 D) AB 160, or (FIG 1 I E) pretreated with 01.2 mg/kg BEV and, 24hr later, AB160.
- FIGs. 11 A-E show in vivo testing of athymic nude mice injected with 1 x 10 A375 human melanoma cells in the right flank and treated with (FIG 11 A) PBS, (FIG 11B) 12 mg/kg BEV, (FIG 1 1 C) 30 mg/kg ABX, (FIG 1 1 D) AB 160, or (FIG 1 I E) pretreated with 01.2 mg/kg BEV and, 24hr later, AB160.
- Data is
- FIG. 1 IF summarizes the day 7-post treatment data from FIGs. 1 1 A-E.
- FIG. 11G summarizes the day 1 0-post treatment data from FIGs. 1 1 A-E.
- FIG. 12 depicts the results of an experiment in which CD20 positive Daudi lymphoma cells were labeled with fluorescent tagged anti-human CD20 or isotype matched control in panels F and A, respectively, and analyzed by flow cytometry.
- the Daudi cells were pretreated with ABRAXANE® (ABX), AR160, AR160L, or Rituxan prior to CD20 labeling.
- ABX ABRAXANE®
- AR160 AR160
- AR160L AR160L
- Rituxan Rituxan
- FIG. 14 depicts a particle size comparison of ABX alone relative to AR and AT freshly made and lyophilized.
- FIG. 15 compares the toxicity of ABX and AR particles in a Daudi cell proliferation assay.
- FIG. 16 depicts the results obtained in mice treated with either labeled
- FIG. 16A depicts the fluorescence accumulation in regions of interest (ROI) in tumor (ROI 2, 3, and 4) and in background areas (ROI 1, 5, and 6). ROI 1, 5 and 6 serve as background references.
- FIG 16B is a bar graph of the average fluorescence per unit of tumor area of mice in all three treatment groups were determined to provide the gross tumor delivery.
- FIG. 16 C is a bar graph of the average fluorescence per unit of tumor area normalized by background ROI to give proportion of drug delivered to tumor versus body. The data demonstrate that administration of AR160 nanoparticles results in an increased fluorescence as compared to ABRAXANE® alone or ABRAXANE® coated with non-specific antibodies.
- FIG. 17 depicts the survival of the mice treated with a single dose of saline, BEV24 (24 mg/kg), ABX30( 30 mg/kg), AB 160 (12mg/kg BEV and 30mg/kg ABX) and AB225 (24 mg/kg BEV and 30 mg/kg ABS).
- BEV24 24 mg/kg
- ABX30 30 mg/kg
- AB 160 (12mg/kg BEV and 30mg/kg ABX
- AB225 24 mg/kg BEV and 30 mg/kg ABS
- compositions and methods include the recited elements, but not excluding others.
- Consisting essentially of when used to define compositions and methods shall mean excluding other elements of any essential significance to the combination for the stated purpose.
- composition consisting essentially of the elements as defined herein would not exclude other materials or steps that do not materially affect the basic and novel characteristic(s) of the claimed invention. "Consisting of shall mean excluding more than trace elements of other ingredients and substantial method steps. Embodiments defined by each of these transition terms are within the scope of this invention.
- nanoparticle refers to particles having at least one dimension which is less than 5 microns. In preferred embodiments, such as for
- the nanoparticle is less than 1 micron.
- the nanoparticle is larger. Even larger particles are expressly contemplated by the invention.
- D50 is the particle size below which 50% of the particles fall. 10%) of particles are smaller than the D IO value and 90% of particles are smaller than D90. Where unclear, the "average" size is equivalent to D50. So, for example, AB160 and AR160 refer to nanoparticles having an average size of 160 nanometers.
- nanoparticle may also encompass discrete multimers of smaller unit nanoparticles.
- a 320 nm particle comprises a dimer of a unit 160 nm nanoparticle.
- multimers would therefore be approximately 320 nm, 480 nm, 640 nm, 800 nm, 960 nm, 1120 nm, and so on.
- carrier protein refers to proteins that function to transport binding agents and/or therapeutic agents.
- the binding agents of the present disclosure can reversibly bind to the carrier proteins. Examples of carrier proteins are discussed in more detail below.
- the term "core” as used herein refers to central or inner portion of the nanoparticle which may be comprised of a carrier protein, a carrier protein and a therapeutic agent, or other agents or combination of agents. In some embodiments, a hydrophobic portion of the binding agent may be incorporated into the core.
- therapeutic agent means an agent which is therapeutically useful, e.g., an agent for the treatment, remission or attenuation of a disease state, physiological condition, symptoms, or etiological factors, or for the evaluation or diagnosis thereof.
- a therapeutic agent may be a chemotherapeutic agent, for example, mitotic inhibitors, topoisomerase inhibitors, steroids, anti-tumor antibiotics,
- binding agent refers to an agent that binds to a target antigen and does not significantly bind to unrelated compounds.
- binding agents that can be effectively employed in the disclosed methods include, but are not limited to, lectins, proteins, and antibodies, such as monoclonal antibodies, e.g. humanized monoclonal antibodies, chimeric antibodies, or polyclonal antibodies, or antigen-binding fragments thereof, as well as aptamers, Fc domain fusion proteins, and aptamers having or fused to hydrophobic protein domain, e.g, Fc domain, etc.
- the binding agent is an exogenous antibody.
- An exogenous antibody is an antibody not naturally produced in a mammal, e.g. in a human, by the mammalian immune system.
- antibody refers to immunoglobulin molecules and immunologically active portions of immunoglobulin molecules (i.e., molecules that contain an antigen binding site that immuno-specifically bind an antigen).
- the term also refers to antibodies comprised of two immunoglobulin heavy chains and two immunoglobulin light chains as well as a variety of forms including full length antibodies and portions thereof; including, for example, an immunoglobulin molecule, a monoclonal antibody, a chimeric antibody, a CDR- grafted antibody, a humanized antibody, a Fab, a Fab', a F(ab')2, a Fv, a disulfide linked Fv, a scFv, a single domain antibody (dAb), a diabody, a multispecific antibody, a dual specific antibody, an anti -idiotypic antibody, a bispecific antibody, a functionally active epitope-binding fragment thereof, bifunctional hybrid antibodies (e.g., Lanzavecchia et al., Eur.
- the antibody may be of any type (e.g., IgG, IgA, IgM, IgE or IgD).
- the antibody is IgG.
- An antibody may be non-human (e.g., from mouse, goat, or any other animal), fully human, humanized, or chimeric.
- Antibody or antibodies include any biosimilar(s) of the antibodies disclosed herein. Biosimilars, as used herein, refers to a biopharmaceutical which is deemed to be comparable in quality, safety, and efficacy to a reference product marketed by an innovator company (Section 351(i) of the Public Health Service Act (42 U.S.C. 262(i)).
- dissociation constant also referred to as "]3 ⁇ 4” refers to a quantity expressing the extent to which a particular substance separates into individual components (e.g., the protein carrier, antibody, and optional therapeutic agent).
- lyophilized refers to a process by which the material (e.g., nanoparticles) to be dried is first frozen and then the ice or frozen solvent is removed by sublimation in a vacuum environment.
- An excipient is optionally included in pre-lyophilized formulations to enhance stability of the lyophilized product upon storage.
- the nanoparticles can be formed from lyophilized components (carrier protein, antibody and optional therapeutic) prior to use as a therapeutic.
- the carrier protein, binding agent, e.g., antibody, and optional therapeutic agent are first combined into nanoparticles and then lyophilized.
- the lyophilized sample may further contain additional excipients.
- bulking agents comprise agents that provide the structure of the fireeze- dried product.
- Common examples used for bulking agents include mannitol, glycine, lactose and sucrose.
- bulking agents may also impart useful qualities in regard to modifying the collapse temperature, providing freeze-thaw protection, and enhancing the protein stability over long-term storage. These agents can also serve as tonicity modifiers.
- buffer encompasses those agents which maintain the solution pH in an acceptable range prior to lyophilization and may include succinate (sodium or potassium), histidine, phosphate (sodium or potassium), Tris(tris(hydroxymethyl)aminom ethane), diethanolamine, citrate (sodium) and the like.
- the buffer of this invention has a pH in the range from about 5.5 to about 6.5; and preferably has a pH of about 6.0. Examples of buffers that will control the pH in this range include succinate (such as sodium succinate), gluconate, histidine, citrate and other organic acid buffers.
- cryoprotectants generally includes agents which provide stability to the protein against freezing-induced stresses, presumably by being preferentially excluded from the protein surface. They may al s o offer protection during primary and secondary drying, and long- term product storage. Examples are polymers such as dextran and polyethylene glycol; sugars such as sucrose, glucose, trehalose, and lactose; surfactants such as polysorbates; and amino acids such as glycine, arginine, and serine.
- lyoprotectanf includes agents that provide stability to the protein during the drying or 'dehydration' process (primary and secondary drying cycles), presumably by providing an amorphous glassy m atri x and by binding with the protein through hydrogen bonding, replacing the water molecules that are removed during the drying process. This helps to maintain the protein conformation, minimize protein degradation during the lyophilization cycle and improve the long-term products.
- examples include polyols or sugars such as sucrose and trehalose.
- composition refers to preparations which are in such form as to permit the active ingredients t o be effective, and which contains no additional components that are toxic to the subjects to which the formulation would be administered.
- “Pharmaceutically acceptable” excipients are those which can reasonably be administered to a subject mammal to provide an effective dose of the active ingredient employed.
- Reconstitution time is the time that is required to rehydrate a lyophilized formulation into a solution.
- a “stable” formulation is one in which the protein therein essentially retains its physical stability and/or chemical stability and/or biological activity upon storage.
- epitope refers to the portion of an antigen which is recognized by a binding agent, e.g., an antibody.
- Epitopes include, but are not limited to, a short amino acid sequence or peptide (optionally glycosylated or otherwise modified) enabling a s p e c i fi c interaction with a protein (e.g., an antibody) or ligand.
- a binding agent e.g., an antibody
- epitopes include, but are not limited to, a short amino acid sequence or peptide (optionally glycosylated or otherwise modified) enabling a s p e c i fi c interaction with a protein (e.g., an antibody) or ligand.
- an epitope may be a part of a molecule to which the antigen-binding site of a binding agent attaches.
- treating covers the treatment of a disease or disorder (e.g., cancer), in a subject, such as a human, and includes: (i) inhibiting a disease or disorder, i.e., arresting its development; (ii) relieving a disease or disorder, i.e., causing regression of the disorder; (iii) slowing progression of the disorder; and/or (iv) inhibiting, relieving, or slowing progression of one or more symptoms of the disease or disorder.
- treating or “treatment” refers to the killing of cancer cells.
- the term "kill" with respect to a cancer treatment is directed to include any type of manipulation that will lead to the death of that cancer cell or at least of portion of a population of cancer cells.
- aptamer refers to a nucleic acid molecule that is capable of binding to a target molecule, such as a polypeptide.
- a target molecule such as a polypeptide.
- an aptamer of the invention can specifically bind to e.g., CD20, CD38, CD52, PD-L1, Ly6E, HER2, HER3/EGFR DAF, ERBB-3 receptor, CSF-1R, STEAP1, CD3, CEA, CD40, OX40, Ang2-VEGF, and VEGF.
- the generation of antibodies with a particular binding specificity and the therapeutic use of aptamers are well e st ab l i s h e d in the art. See, e.g., U.S. Pat. No.
- Fc-fusion proteins are bioengineered polypeptides that join the crystallizable fragment (Fc) domain of an antibody with another biologically active agent, e.g., a protein domain, peptide, o r nucleic acid or peptide aptamer to generate a molecule with desired structure- function properties and significant therapeutic potential.
- the gamma immunoglobulin (IgG) isotype is often used as the basis for generating Fc-fusion proteins because of favorable characteristics such as recruitment of effector function and increased plasma half-life.
- Fc- fusion proteins Given the range of aptamers, both peptide and nucleic acids, that can be used as fusion partners, Fc- fusion proteins have numerous biological and pharmaceutical applications.
- the current invention is predicated, in part, on the surprising discovery that optionally lyophilized nanoparticles comprising a carrier protein, a binding agent, e.g., an antibody, an aptamer, or a fusion protein having a hydrophobic domain and a binding domain, e.g., an Fc domain fused to an aptamer or the ligand of a cellular receptor, and a therapeutic agent provide targeted therapy to a tumor while minimizing toxicity to the patient.
- a binding agent e.g., an antibody, an aptamer, or a fusion protein having a hydrophobic domain and a binding domain, e.g., an Fc domain fused to an aptamer or the ligand of a cellular receptor
- a therapeutic agent provide targeted therapy to a tumor while minimizing toxicity to the patient.
- ADCs Another shortcoming of current ADCs is that higher drug penetration into the tumor has not been substantively proven in human tumors.
- Early testing of ADCs in mouse models suggested that tumor targeting with antibodies would result in a higher concentration of the active agent in the tumor (Deguchi, T. et al. (1986) Cancer Res 46: 3751-3755); however, this has not correlated in the treatment of human disease, likely because human tumors are much more heterogeneous in permeability than mouse tumors. Jain, R.K. et al. (2010) Nat Rev Clin Oncol 7:653-664. Also, the size of the nanoparticle is critical for extravasation from the vasculature into the tumor.
- the nano-immune conjugate described herein overcomes this issue by the fact that the large complex, which is less than 200 nm intact, is partially dissociated in systemic circulation into smaller functional units that are easily able to permeate tumor tissue. Furthermore, once the conjugate arrives to the tumor site, the smaller toxic payload can be released and only the toxic portion needs to be taken up by tumor cells, not the entire conjugate.
- compositions of albumin and an binding agent e.g., antibody
- an binding agent e.g., antibody
- the binding agents useful in this invention spontaneously self-assemble into and onto the albumin to form nanoparticles having multiple copies of the binding agent (up to 500 or more).
- the antigen (or ligand) receptor portion of the binding agent e.g., the antibody or aptamer or Fc fusion molecule, is positioned outward from the nanoparticle while the hydrophobic tail of the binding agent in integrated into the albumin by hydrophobic - hydrophobic interactions.
- ionic charges on the proteins are dehydrated thereby exposing the underlying charges. Exposed charges allow for charge- charge interactions between the two proteins which can alter the binding affinity of each protein to the other.
- concentration of the nanoparticles increases significantly as the solvent (e.g., water) is removed. Such increased concentrations of nanoparticles could lead to irreversible oligomerization.
- Oligomerization is a known property of proteins that reduces the biological properties of the oligomer as compared to the monomelic form and increases the size of the particle sometimes beyond 1 micron.
- a stable form of a nanoparticle composition is required for clinical and/or commercial use where a shelf-life of at least 3 months is required and shelf- lives of greater than 6 months or 9 months are preferred.
- Such a stable composition must be readily available for intravenous injection, must retain its self-assembled form upon intravenous injection so as to direct the nanoparticle to the predetermined site in vivo, must have a maximum size of less than 1 micron so as to avoid any ischemic event when delivered into the blood stream, and finally must be compatible with the aqueous composition used for injection.
- compositions of nanoparticles containing a carrier protein, binding agents, and optionally at least one therapeutic agent wherein said compositions are optionally lyophilized.
- the carrier protein can be albumin, gelatin, elastin (including topoelastin) or elastin-derived polypeptides (e.g., a-elastin and elastin-like polypeptides (ELPs)), gliadin, legumin, zein, soy protein (e.g., soy protein isolate (SPI)), milk protein (e.g., ⁇ -lactoglobulin (BLG) and casein), or whey protein (e.g., whey protein concentrates (WPC) and whey protein isolates (WPI)).
- the carrier protein is albumin.
- the albumin is egg white (ovalbumin), bovine serum albumin (BSA), or the like.
- the carrier protein is human serum albumin (HSA).
- the carrier protein is a generally regarded as safe (GRAS) excipient approved by the United States Food and Drug
- the binding agents are antibodies selected from the group consisting of ado- trastuzumab emtansine, alemtuzumab, bevacizumab, cetuximab, denosumab, dinutuximab, ipilimumab, nivolumab, obinutuzumab, ofatumumab, panitumumab, pembrolizumab, pertuzumab, rituximab, and trastuzumab.
- the antibodies are a substantially single layer of antibodies on all or part of the surface of the nanoparticle.
- Table 1 depicts a list of non-limiting list of antibodies.
- Table 1 Antibodies
- RG7221 (Ang2-VEGF mAb) Metastatic colorectal cancer
- RG7686 (glypican-3 mAb) Hepatocellular carcinoma
- the at least one therapeutic agent is selected from the group consisting of abiraterone, bendamustine, bortezomib, carboplatin, cabazitaxel, cisplatin, chlorambucil, dasatinib, docetaxel, doxorubicin, epirubicin, erlotinib, etoposide, everolimus, gefitinib, idarubicin, imatinib, hydroxyurea, imatinib, lapatinib, leuprorelin, melphalan, methotrexate, mitoxantrone, nedaplatin, nilotinib, oxaliplatin, paclitaxel, pazopanib, pemetrexed, picoplatin, romidepsin, satraplatin, sorafenib, vemurafenib, sunitinib, tenipos
- Table 2 depicts a list of non-limiting list of cancer therapeutic agents.
- Adriamycin Doxorubicin Hydrochloride
- Acute lymphoblastic leukemia acute myeloid leukemia; breast cancer, gastric (stomach) cancer; Hodgkin lymphoma; neuroblastoma; non-Hodgkin lymphoma; ovarian cancer; small cell lung cancer; soft tissue and bone sarcomas; thyroid cancer; transitional cell bladder cancer; Wilms tumor
- Adrucil Basal cell carcinoma; breast cancer;
- colorectal cancer gastric (stomach) adenocarcinoma
- pancreatic cancer squamous cell carcinoma of the head and neck
- Alimta (Pemetrexed Disodium) Malignant pleural mesothelioma; non- small cell lung cancer
- Ambochlorin Chronic lymphocytic leukemia
- Aromasin (Exemestane) Advanced breast cancer early-stage
- Arranon (Nelarabine) T-cell acute lymphoblastic leukemia; T- cell lymphoblastic lymphoma
- BiCNU Carmustine Brain tumors; Hodgkin lymphoma;
- lymphoma penile cancer
- squamous cell carcinoma of the cervix squamous cell carcinoma of the head and neck
- squamous cell carcinoma of the vulva testicular cancer
- Busulfex (Busulfan) Chronic myelogenous leukemia
- Cerubidine (Daunorubicin Hydrochloride) Acute lymphoblastic leukemia; acute myeloid leukemia
- Cisplatin Bladder cancer cervical cancer; malignant mesothelioma; non-small cell lung cancer; ovarian cancer; squamous cell carcinoma of the head and neck; testicular cancer
- Clafen (Cyclophosphamide) Acute lymphoblastic leukemia; acute
- myeloid leukemia breast cancer; chronic lymphocytic leukemia; chronic myelogenous leukemia; Hodgkin lymphoma; multiple myeloma; mycosis fungoides;
- neuroblastoma neuroblastoma
- non- Hodgkin lymphoma ovarian cancer
- Clofarex (Clofarabine) Acute lymphoblastic leukemia
- Cosmegen (Dactinomycin) Ewing sarcoma; gestational trophoblastic disease; rhabdomyosarcoma; solid tumors; testicular cancer; Wilms tumor
- Cyclophosphamide Acute lymphoblastic leukemia; acute
- myeloid leukemia breast cancer; chronic lymphocytic leukemia; chronic myelogenous leukemia; Hodgkin lymphoma; multiple myeloma; mycosis fungoides;
- neuroblastoma neuroblastoma
- non- Hodgkin lymphoma ovarian cancer
- retinoblastoma retinoblastoma
- Cyramza (Ramucirumab) Adenocarcinoma; colorectal cancer; non- small cell lung cancer
- myeloid leukemia chronic myelogenous leukemia; meningeal leukemia
- Cytosar-U (Cytarabine) Acute lymphoblastic leukemia; acute
- myeloid leukemia chronic myelogenous leukemia
- meningeal leukemia Cytoxan Cyclophosphamide
- Acute lymphoblastic leukemia acute myeloid leukemia
- breast cancer chronic lymphocytic leukemia
- chronic myelogenous leukemia myeloid leukemia
- myeloma mycosis fungoides
- lymphoma ovarian cancer
- Daunorubicin Hydrochloride Acute lymphoblastic leukemia; acute
- stomach or gastroesophageal junction non- small cell lung cancer; prostate cancer;
- Doxil Doxorubicin Hydrochloride Liposome
- Doxorubicin Hydrochloride Acute lymphoblastic leukemia; acute myeloid leukemia; breast cancer; gastric (stomach) cancer; Hodgkin lymphoma; neuroblastoma; non-Hodgkin lymphoma; ovarian cancer; small cell lung cancer; soft tissue and bone sarcomas; thyroid cancer; transitional cell bladder cancer; Wilms tumor.
- Dox-SL Doxorubicin AIDS-related Kaposi sarcoma
- DTIC-Dome Hodgkin lymphoma; melanoma
- Efudex Basal cell carcinoma; breast cancer;
- colorectal cancer gastric (stomach) adenocarcinoma
- pancreatic cancer squamous cell carcinoma of the head and neck
- Eloxatin (Oxaliplatin) Colorectal cancer; stage III colon cancer
- Erbitux Colorectal cancer; squamous cell carcinoma of the head and neck Eribulin Mesylate Breast cancer
- Etopophos Small cell lung cancer; testicular cancer
- Fludara (Fludarabine Phosphate) Chronic lymphocytic leukemia
- Fluoroplex Basal cell carcinoma; breast cancer;
- colorectal cancer gastric (stomach) adenocarcinoma
- pancreatic cancer squamous cell carcinoma of the head and neck
- gestational trophoblastic disease head and neck cancer
- lung cancer mycosis fungoides
- non-Hodgkin lymphoma non-Hodgkin lymphoma
- FU-LV Colorectal cancer esophageal cancer
- cervical cancer malignant mesothelioma; non-small cell lung cancer; ovarian cancer; pancreatic cancer
- Gemzar (Gemcitabine Hydrochloride) Breast cancer; non-small cell lung
- Gleevec (Imatinib Mesylate) Acute lymphoblastic leukemia; chronic myeloblastic leukemia
- eosinophilic leukemia or hypereosinophilic syndrome eosinophilic leukemia or hypereosinophilic syndrome
- chronic myelogenous leukemia eosinophilic leukemia or hypereosinophilic syndrome
- dermatofibrosarcoma protuberans eosinophilic leukemia or hypereosinophilic syndrome
- chronic myelogenous leukemia eosinophilic leukemia or hypereosinophilic syndrome
- chronic myelogenous leukemia dermatofibrosarcoma protuberans
- myelodysplastic/myeloproliferative neoplasms myelodysplastic/myeloproliferative neoplasms; systemic mastocytosis.
- Gliadel Carmustine Implant
- Glioblastoma multiforme malignant glioma
- Hycamtin Topotecan Hydrochloride Cervical cancer; ovarian cancer; small cell lung cancer
- Hyper-CVAD Acute lymphoblastic leukemia; non- Hodgkin lymphoma
- Iclusig Ponatinib Hydrochloride
- Idamycin (Idarubicin Hydrochloride) Acute myeloid leukemia
- Imatinib Mesylate Acute lymphoblastic leukemia; chronic
- eosinophilic leukemia or hypereosinophilic syndrome eosinophilic leukemia or hypereosinophilic syndrome
- chronic myelogenous leukemia eosinophilic leukemia or hypereosinophilic syndrome
- dermatofibrosarcoma protuberans eosinophilic leukemia or hypereosinophilic syndrome
- chronic myelogenous leukemia eosinophilic leukemia or hypereosinophilic syndrome
- chronic myelogenous leukemia dermatofibrosarcoma protuberans
- myelodysplastic/myeloproliferative neoplasms myelodysplastic/myeloproliferative neoplasms; systemic mastocytosis.
- Imbruvica (Ibrutinib) Chronic lymphocytic leukemia; mantle cell lymphoma; Waldenstr6m
- Linfolizin Chlorambucil
- Chronic lymphocytic leukemia Chronic lymphocytic leukemia
- LipoDox Doxorubicin AIDS-related Kaposi sarcoma
- lymphocytic leukemia chronic myelogenous leukemia
- Hodgkin lymphoma malignant pleural effusion, malignant pericardial effusion, and malignant peritoneal effusion
- mycosis fungoides non-Hodgkin lymphoma
- Methazolastone (Temozolomide) Anaplastic astrocytoma
- Mexate Acute lymphoblastic leukemia
- gestational trophoblastic disease head and neck cancer
- lung cancer mycosis fungoides
- non-Hodgkin lymphoma non-Hodgkin lymphoma
- Mexate-AQ Acute lymphoblastic leukemia
- gestational trophoblastic disease head and neck cancer
- lung cancer mycosis fungoides
- non-Hodgkin lymphoma non-Hodgkin lymphoma
- Mitozytrex (Mitomycin C) Gastric (stomach) and
- lymphocytic leukemia chronic myelogenous leukemia
- Hodgkin lymphoma malignant pleural effusion, malignant pericardial effusion, and malignant peritoneal effusion
- mycosis fungoides non-Hodgkin lymphoma
- Mylotarg (Gemtuzumab Ozogamicin) Acute myeloid leukemia
- Nanoparticle Paclitaxel (Paclitaxel Albumin- Breast cancer; Non-small cell lung stabilized Nanoparticle Formulation) cancer; Pancreatic cancer
- Neosar (Cyclophosphamide) Acute lymphoblastic leukemia; Acute
- myeloid leukemia Breast cancer; Chronic lymphocytic leukemia; Chronic myelogenous leukemia; Hodgkin lymphoma; Multiple myeloma; Mycosis fungoides;
- Neuroblastoma Non- Hodgkin lymphoma; Ovarian cancer; Retinoblastoma Nexavar (Sorafenib Tosylate) Hepatocellular carcinoma; Renal
- Nivolumab Melanoma Squamous non-small cell
- Nolvadex (Tamoxifen Citrate) Breast cancer
- Non-small cell lung cancer Ovarian
- Platinol (Cisplatin) Bladder cancer Cervical cancer; Malignant mesothelioma; Non-small cell lung cancer; Ovarian cancer; Squamous cell carcinoma of the head and neck; Testicular cancer
- Platinal-AQ (Cisplatin) Bladder cancer; Cervical cancer; Malignant mesothelioma; Non-small cell lung cancer; Ovarian cancer; Squamous cell carcinoma of the head and neck; Testicular cancer
- Prednisone Acute lymphoblastic leukemia; Chronic lymphocytic leukemia; Hodgkin lymphoma; Multiple myeloma; Non-Hodgkin lymphoma; Prostate cancer; Thymoma and thymic carcmoma
- Provenge (Sipuleucel-T) Prostate cancer Purinethol (Mercaptopurine) Acute lymphoblastic leukemia
- Rheumatrex (Methotrexate) Acute lymphoblastic leukemia
- Rubidomycin Daunorubicin Hydrochloride Acute lymphoblastic leukemia; Acute
- Somatuline Depot (Lanreotide Acetate) Gastroenteropancreatic neuroendocrine tumors
- Sprycel Acute lymphoblastic leukemia
- Tasigna Chronic myelogenous leukemia
- Taxol (Paclitaxel) AIDS-related Kaposi sarcoma; Breast
- Non-small cell lung cancer Ovarian
- Taxotere Docetaxel Breast cancer; Adenocarcinoma; Non- small cell lung cancer; Prostate cancer; Squamous cell carcinoma of the head and
- Thiotepa Bladder cancer ; Breast cancer; Malignant pleural effusion, malignant pericardial effusion, and malignant peritoneal effusion; Ovarian cancer
- Torisel (Temsirolimus) Renal cell carcinoma
- TPF Squamous cell carcinoma of the head and neck; Gastric (stomach) cancer
- Treanda Bendamustine Hydrochloride B-cell non-Hodgkin lymphoma
- Trisenox Arsenic Trioxide Acute promyelocytic leukemia
- Velban (Vinblastine Sulfate) Breast cancer; Choriocarcinoma;
- Velsar Vehicle Sulfate Breast cancer; Choriocarcinoma;
- Hodgkin lymphoma Kaposi sarcoma; Mycosis fungoides; Non-Hodgkin lymphoma; Testicular cancer
- VePesid Small cell lung cancer
- Vincasar PFS Vincristine Sulfate Acute leukemia; Hodgkin lymphoma;
- Zydelig Chronic lymphocytic leukemia; Non- Hodgkin lymphoma (Follicula B-cell non Hodgkin lymphoma and Small lymphocytic
- the therapeutic agent may be located inside the nanoparticle, on the outside surface of the nanoparticle, or both.
- the nanoparticle may contain more than one therapeutic agent, for example, two therapeutic agents, three therapeutic agents, four therapeutic agents, five therapeutic agents, or more.
- a nanoparticle may contain the same or different therapeutic agents inside and outside the nanoparticle.
- nanoparticles comprising ABRAXA E and bevacizumab are excluded.
- the nanoparticle comprises at least 100 binding agents non-covalently bound to the surface of the nanoparticle. In one aspect, the nanoparticle comprises at least 200 binding agents non-covalently bound to the surface of the nanoparticle. In one aspect, the nanoparticle comprises at least 300 binding agents non-covalently bound to the surface of the nanoparticle. In one aspect, the nanoparticle comprises at least 400 binding agents non-covalently bound to the surface of the nanoparticle. In one aspect, the nanoparticle comprises at least 500 binding agents non-covalently bound to the surface of the nanoparticle. In one aspect, the nanoparticle comprises at least 600 binding agents non- covalently bound to the surface of the nanoparticle.
- the nanoparticle comprises between about 100 and about 1000 binding agents non-covalently bound to the surface of the nanoparticle. In one aspect, the nanoparticle comprises between about 200 and about 1000 binding agents non-covalently bound to the surface of the nanoparticle. In one aspect, the nanoparticle comprises between about 300 and about 1000 binding agents non-covalently bound to the surface of the nanoparticle. In one aspect, the nanoparticle comprises between about 400 and about 1000 binding agents non-covalently bound to the surface of the nanoparticle. In one aspect, the nanoparticle comprises between about 500 and about 1000 binding agents non- covalently bound to the surface of the nanoparticle.
- the nanoparticle comprises between about 600 and about 1000 binding agents non-covalently bound to the surface of the nanoparticle. In one aspect, the nanoparticle comprises between about 200 and about 800 binding agents non-covalently bound to the surface of the nanoparticle. In one aspect, the nanoparticle comprises between about 300 and about 800 binding agents non-covalently bound to the surface of the nanoparticle. In preferred embodiments, the nanoparticle comprises between about 400 and about 800 binding agents non-covalently bound to the surface of the nanoparticle. Contemplated values include any value or subrange within any of the recited ranges, including endpoints.
- the average particle size in the nanoparticle composition is less than about 1 ⁇ . In one aspect, the average particle size in the nanoparticle composition is between about 130 nm and about 1 ⁇ . In one aspect, the average particle size in the nanoparticle composition is between about 130 nm and about 900 nm. In one aspect, the average particle size in the nanoparticle composition is between about 130 nm and about 800 nm. In one aspect, the average particle size in the nanoparticle composition is between about 130 nm and about 700 nm. In one aspect, the average particle size in the
- nanoparticle composition is between about 130 nm and about 600 nm. In one aspect, the average particle size in the nanoparticle composition is between about 130 nm and about 500 nm. In one aspect, the average particle size in the nanoparticle composition is between about 130 nm and about 400 nm. In one aspect, the average particle size in the
- nanoparticle composition is between about 130 nm and about 300 nm. In one aspect, the average particle size in the nanoparticle composition is between about 130 nm and about 200 nm. In a preferred embodiment, the average particle size in the nanoparticle composition is between about 150 nm and about 180 nm. In an especially preferred embodiment, the mean particle size in the nanoparticle composition is about 160 nm. Contemplated values include any value, subrange, or range within any of the recited ranges, including endpoints.
- the nanoparticle composition is formulated for intravenous injection.
- the nanoparticle composition formulated for intravenous injection should comprise nanoparticles with an average particle size of less than about 1 ⁇ .
- the average particle size in the nanoparticle composition is greater than about 1 ⁇ . In one aspect, the average particle size in the nanoparticle composition is between about 1 ⁇ and about 5 ⁇ . In one aspect, the average particle size in the nanoparticle composition is between about 1 ⁇ and about 4 ⁇ . In one aspect, the average particle size in the nanoparticle composition is between about 1 ⁇ and about 3 ⁇ . In one aspect, the average particle size in the nanoparticle composition is between about 1 ⁇ and about 2 ⁇ . In one aspect, the average particle size in the nanoparticle composition is between about 1 ⁇ and about 1.5 ⁇ . Contemplated values include any value, subrange, or range within any of the recited ranges, including endpoints.
- the nanoparticle composition is formulated for direct injection into a tumor.
- Direct injection includes injection into or proximal to a tumor site, perfusion into a tumor, and the like.
- the nanoparticle may comprise any average particle size. Without being bound by theory, it is believed that larger particles (e.g., greater than 500 nm, greater than 1 ⁇ , and the like) are more likely to be immobilized within the tumor, thereby providing a beneficial effect. Larger particles can accumulate in the tumor or specific organs. See, e.g., 20-60 micron glass particle that is used to inject into the hepatic artery feeding a tumor of the liver, called "TheraSphere®" (in clinical use for liver cancer). Therefore, for intravenous administration, particles under 1 ⁇ are typically used. Particles over 1 ⁇ are, more typically, administered directly into a tumor (“direct injection”) or into an artery feeding into the site of the tumor.
- less than about 0.01% of the nanoparticles within the composition have a particle size greater than 200 nm, greater than 300 nm, greater than 400 nm, greater than 500 nm, greater than 600 nm, greater than 700 nm, or greater than 800 nm. In one aspect, less than about 0.001% of the nanoparticles within the composition have a particle size greater than 200 nm, greater than 300 nm, greater than 400 nm, greater than 500 nm, greater than 600 nm, greater than 700 nm, or greater than 800 nm. In a preferred embodiment, less than about 0.01% of the nanoparticles within the composition have a particle size greater than 800 nm.
- the sizes and size ranges recited herein relate to particle sizes of the reconstituted lyophilized nanoparticle composition. That is, after the lyophilized nanoparticles are resuspended in an aqueous solution (e.g., water, other pharmaceutically acceptable excipient, buffer, etc.), the particle size or average particle size is within the range recited herein.
- an aqueous solution e.g., water, other pharmaceutically acceptable excipient, buffer, etc.
- At least about 50%, 60%, 70%, 80%, 90%, 95%, 96%, 97%, 98%, 99%), 99.5%), or 99.9% of the nanoparticles are present in the reconstituted composition as single nanoparticles. That is, fewer than about 50%, 40%, 30%, etc. of the nanoparticles are dimerized or multimerized (oligomerized).
- the size of the nanoparticle can be controlled by the adjusting the amount (e.g., ratio) of carrier protein to binding agent.
- the size of the nanoparticles, and the size distribution, is also important.
- the nanoparticles of the invention may behave differently according to their size. At large sizes, an agglomeration may block blood vessels. Therefore, agglomeration of nanoparticles can affect the performance and safety of the composition. On the other hand, larger particles may be more therapeutic under certain conditions (e.g., when not administered intravenously).
- the nanoparticle composition comprises at least one additional therapeutic agent.
- the at least one additional therapeutic agent is non- covalently bound to the outside surface of the nanoparticle.
- the at least one additional therapeutic agent is arranged on the outside surface of the nanoparticle.
- the at least one additional therapeutic agent is selected from the group consisting of abiraterone, bendamustine, bortezomib, carboplatin, cabazitaxel, cisplatin, chlorambucil, dasatinib, docetaxel, doxorubicin, epirubicin, erlotinib, etoposide, everolimus, gemcitabine, gefitinib, idarubicin, imatinib, hydroxyurea, imatinib, lapatinib, leuprorelin, melphalan, methotrexate, mitoxantrone, nedaplatin, nilotinib, oxaliplatin, pazopanib, pemetrexed, picoplatin, romidepsin, satraplatin, sorafenib, vemurafenib, sunitinib, teniposide, triplatin, vin
- the current invention relates to methods of making nanoparticle compositions as described herein.
- the nanoparticles of the nanoparticle composition are formed by contacting the carrier protein or carrier protein-therapeutic agent particle with the binding agent at a ratio of about 10: 1 to about 10:30 carrier protein particle or carrier protein- therapeutic agent particle to binding agent.
- the ratio is about 10:2 to about 10:25.
- the ratio is about 10:2 to about 1 : 1.
- the ratio is about 10:2 to about 10:6.
- the ratio is about 10:4.
- Contemplated ratios include any value, subrange, or range within any of the recited ranges, including endpoints.
- the amount of solution or other liquid medium employed to form the nanoparticles is particularly important. No nanoparticles are formed in an overly dilute solution of the carrier protein (or carrier protein-therapeutic agent) and the antibodies. An overly concentrated solution will result in unstructured aggregates.
- the amount of solution e.g., sterile water, saline, phosphate buffered saline
- the amount of carrier protein is between about 1 mg/mL and about lOO mg/mL.
- the amount of binding agent is between about 1 mg/mL and about 30 mg/mL.
- the ratio of carrier proteimbinding agent: solution is approximately 9 mg of carrier protein (e.g., albumin) to 4 mg of binding agent, e.g., antibody (e.g., BEV) in 1 mL of solution (e.g., saline).
- An amount of therapeutic agent e.g., taxol
- 1 mg of taxol can be added 9 mg of carrier protein (10 mg carrier protein- therapeutic) and 4 mg of binding agent, e.g., antibody, Fc fusion molecule, or aptamer, in 1 mL of solution.
- binding agent e.g., antibody, Fc fusion molecule, or aptamer
- the carrier protein or carrier protein-therapeutic agent particle is contacted with the binding agent in a solution having a pH between about 4 and about 8. In one embodiment, the carrier protein or carrier protein-therapeutic agent particle is contacted with the binding agent in a solution having a pH of about 4. I n one embodiment, the carrier protein or carrier protein-therapeutic agent particle is contacted with the binding agent in a solution having a pH of about 5. In one embodiment, the carrier protein or carrier protein-therapeutic agent particle is contacted with the binding agent in a solution having a pH of about 6. In one embodiment, the carrier protein or carrier protein- therapeutic agent particle is contacted with the binding agent in a solution having a pH of about 7.
- the carrier protein or carrier protein-therapeutic agent particle is contacted with the binding agent in a solution having a pH of about 8. In a preferred embodiment, the carrier protein or carrier protein-therapeutic agent particle is contacted with the binding agent in a solution having a pH between about 5 and about 7.
- the carrier protein particle or carrier protein-therapeutic agent particle is incubated with the binding agent at a temperature of about 5 °C to about 60 °C, or any range, subrange, or value within that range including endpoints. In a preferred embodiment, the carrier protein particle or carrier protein-therapeutic agent particle is incubated with the binding agent at a temperature of about 23 °C to about 60 °C.
- the stability of the nanoparticles within the nanoparticle composition is, at least in part, dependent upon the temperature and/or pH at which the nanoparticles are formed, as well as the concentration of the components (i.e., carrier protein, binding agent, and optionally therapeutic agent) in the solution.
- the K d of the nanoparticles is between about 1 x 10 "11 M and about 2 x 10 "5 M. In one embodiment, the K d of the nanoparticles is between about 1 x 10 "11 M and about 2 x 10 "8 M. In one embodiment, the K d of the nanoparticles is between about 1 x 10 "11 M and about 7 x 10 "9 M.
- the K d of the nanoparticles is between about 1 x 1 0 " 1 1 M and about 3x 10*M.
- Contemplated values include any value, subrange, or range within any of the recited ranges, including endpoints.
- compositions of this invention are prepared by standard
- nanoparticles retain their size distribution upon lyophilization and, more importantly, can be reconstituted for in vivo administration (e.g., intravenous delivery) in substantially the same form and ratios as if freshly made.
- the nanoparticle composition is formulated for systemic delivery, e.g., intravenous administration.
- the nanoparticle composition is formulated for direct injection into a tumor.
- Direct injection includes injection into or proximal to a tumor site, perfusion into a tumor, and the like. Because the nanoparticle composition is not administered
- a nanoparticle composition is formulated for direct injection into a tumor may comprise any average particle size. Without being bound by theory, it is believed that larger particles (e.g., greater than 500 nm, greater than 1 ⁇ , and the like) are more likely to be immobilized within the tumor, thereby providing what is believed to be a better beneficial effect.
- composition comprising a compound provided herein, and at least one pharmaceutically acceptable excipient.
- the compounds provided herein can be formulated for administration to a patient by any of the accepted modes of administration.
- Various formulations and drug delivery systems are available in the art. See, e.g., Gennaro, A.R., ed. (1995) Remington's Pharmaceutical Sciences, 18th ed., Mack Publishing Co.
- compounds provided herein will be administered as pharmaceutical compositions by any one of the following routes: oral, systemic (e.g., transdermal, intranasal or by suppository), or parenteral (e.g., intramuscular, intravenous or
- compositions are comprised of, in general, a compound of the present invention in combination with at least one pharmaceutically acceptable excipient.
- excipients are non-toxic, aid administration, and do not adversely affect the therapeutic benefit of the claimed compounds.
- excipient may be any solid, liquid, semi-solid or, in the case of an aerosol composition, gaseous excipient that is generally available to one of skill in the art.
- Solid pharmaceutical excipients include starch, cellulose, talc, glucose, lactose, sucrose, gelatin, malt, rice, flour, chalk, silica gel, magnesium stearate, sodium stearate, glycerol monostearate, sodium chloride, dried skim milk and the like.
- Liquid and semisolid excipients may be selected from glycerol, propylene glycol, water, ethanol and various oils, including those of petroleum, animal, vegetable or synthetic origin, e.g., peanut oil, soybean oil, mineral oil, sesame oil, etc.
- Preferred liquid carriers, particularly for injectable solutions include water, saline, aqueous dextrose, and glycols.
- Other suitable pharmaceutical excipients and their formulations are described in Remington's
- compositions may, if desired, be presented in a pack or dispenser device containing one or more unit dosage forms containing the active ingredient.
- a pack or device may, for example, comprise metal or plastic foil, such as a blister pack, or glass, and rubber stoppers such as in vials.
- the pack or dispenser device may be accompanied by instructions for administration.
- Compositions comprising a compound of the invention formulated in a compatible pharmaceutical carrier may also be prepared, placed in an appropriate container, and labeled for treatment of an indicated condition.
- the nanoparticle compositions as described herein are useful in treating cancer cells and/or tumors in a mammal.
- the mammal is a human (i.e., a human patient).
- the lyophilized nanoparticle composition is
- Treatment of a cancer cell includes, without limitation, reduction in proliferation, killing the cell, preventing metastasis of the cell, and the like.
- a method for treating a tumor in a patient in need thereof comprising administering to the patient a therapeutically effective amount of a nanoparticle composition as described herein to treat the tumor.
- the size of the tumor is reduced.
- the tumor size does not increase (i.e. progress) for at least a period of time during and/or after treatment.
- the nanoparticle composition is administered intravenously. In one embodiment, the nanoparticle composition is administered directly to the tumor. In one embodiment, the nanoparticle composition is administered by direct injection or perfusion into the tumor.
- the method comprises: a) administering the nanoparticle composition once a week for three weeks; b) ceasing administration of the nanoparticle composition for one week; and c) optionally repeating steps a) and b) as necessary to treat the tumor.
- the therapeutically effective amount of the nanoparticles described herein comprises about 50 mg/m 2 to about 200 mg/m 2 carrier protein or carrier protein and therapeutic agent. In a preferred embodiment, the therapeutically effective amount comprises about 75 mg/m 2 to about 175 mg/m 2 carrier protein or carrier protein and therapeutic agent.
- Contemplated values include any value, subrange, or range within any of the recited ranges, including endpoints.
- the therapeutically effective amount comprises about 20 mg/m 2 to about 90 mg/m 2 binding agent, e.g., antibody, aptamer or Fc fusion. In a preferred embodiment, the therapeutically effective amount comprises 30 mg/m 2 to about 70 mg/m 2 binding agent, e.g., antibody, aptamer or Fc fusion.
- Contemplated values include any value, subrange, or range within any of the recited ranges, including endpoints.
- Cancers or tumors that can be treated by the compositions and methods described herein include, but are not limited to: biliary tract cancer; brain cancer, including glioblastomas and medulloblastomas; breast cancer; cervical cancer; choriocarcinoma; colon cancer; endometrial cancer; esophageal cancer, gastric cancer; hematological neoplasms, including acute lymphocytic and myelogenous leukemia; multiple myeloma; AIDS associated leukemias and adult T-cell leukemia lymphoma; intraepithelial neoplasms, including Bowen's disease and Paget's disease; liver cancer (hepatocarcinoma); lung cancer; lymphomas, including Hodgkin's disease and lymphocytic lymphomas; neuroblastomas; oral cancer, including squamous cell carcinoma; ovarian cancer, including those arising from epithelial cells, stromal cells, germ cells and mesenchymal
- pancreas cancer pancreas cancer
- prostate cancer rectal cancer
- sarcomas including leiomyosarcoma, rhabdomyosarcoma, liposarcoma, fibrosarcoma and osteosarcoma
- skin cancer including melanoma, Kaposi's sarcoma, basocellular cancer and squamous cell cancer
- testicular cancer including germinal tumors (seminoma, non-seminoma[teratomas,
- cancers or tumors include breast cancer, lymphoma, multiple myeloma, and melanoma.
- the compound of this invention i.e., the nanoparticles
- the actual amount of the compound of this invention will depend upon numerous factors such as the severity of the disease to be treated, the age and relative health of the subject, the potency of the compound used, the route and form of administration, and other factors well known to the skilled artisan.
- an effective amount or a therapeutically effective amount or dose of an agent refers to that amount of the agent or compound that results in amelioration of symptoms or a prolongation of survival in a subject.
- Toxicity and therapeutic efficacy of such molecules can be determined by standard pharmaceutical procedures in cell cultures or experimental animals, e.g., by determining the LD50 (the dose lethal to 50% of the population) and the ED50 (the dose therapeutically effective in 50% of the population).
- the dose ratio of toxic to therapeutic effects is the therapeutic index, which can be expressed as the ratio LD50/ED50. Agents that exhibit high therapeutic indices are preferred.
- the effective amount or therapeutically effective amount is the amount of the compound or pharmaceutical composition that will elicit the biological or medical response of a tissue, system, animal or human that is being sought by the researcher, veterinarian, medical doctor or other clinician. Dosages may vary within this range depending upon the dosage form employed and/or the route of administration utilized. The exact formulation, route of administration, dosage, and dosage interval should be chosen according to methods known in the art, in view of the specifics of a subject's condition.
- Dosage amount and interval may be adjusted individually to provide plasma levels of the active moiety that are sufficient to achieve the desired effects; i.e., the minimal effective concentration (MEC).
- MEC minimal effective concentration
- the MEC will vary for each compound but can be estimated from, for example, in vitro data and animal experiments. Dosages necessary to achieve the MEC will depend on individual characteristics and route of administration. In cases of local administration or selective uptake, the effective local concentration of the drug may not be related to plasma concentration.
- nanoparticles composed of albumin- bound paclitaxel (i.e., ABRAXA E®) or cisplatin as core, and bevacizumab (i.e., Avastin®) or Rituximab (i.e., Rituxan®) as antibodies.
- ADC antibody dependent chemotherapy
- ABRAXA E® (ABX) (10 mg) was suspended in bevacizumab (BEV) (4 mg [160 ⁇ ] unless otherwise indicated), and 840 ⁇ of 0.9% saline was added to give a final concentration of 10 mg/ml and 2 mg/ml of ABX and BEV, respectively. The mixture was incubated for 30 minutes at room temperature (or at the temperature indicated) to allow particle formation. For Mastersizer experiments to measure particle size of ABX:BEV complexes, 10 mg of ABX was suspended in BEV at concentrations of 0 to 25 mg/ml. Complexes of ABX with rituximab (0-10 mg/ml) or trastuzumab (0-22 mg/ml) were formed under similar conditions.
- the ABX:BEV complexes may be prepared by obtaining the dose appropriate number of 4 mL vials of 25 mg/mL BEV and diluting each vial per the following directions to 4 mg/mL.
- the dose appropriate number of 100 mg vials of ABX can be prepared by reconstituting to a final concentration containing 10 mg/mL ABX nanoparticles.
- 1.6 mL (40 mg) of bevacizumab (25 mg/mL) can be withdrawn and slowly injected, over a minimum of 1 minute, onto the inside wall of each of the vials containing 100 mg of ABX.
- the bevacizumab solution should not be injected directly onto the lyophilized cake as this will result in foaming. Then, using a sterile 12 mL sterile syringe, 8.4 mL 0.9% Sodium Chloride Injection, USP, can be withdrawn and slowly injected, over a minimum of 1 minute, 8.4 mL onto the inside wall of each vial containing ABX 100 mg and BEV 40 mg. Once the addition of BEV 1.6 mL and 0.9% Sodium Chloride Injection, USP 8.4 mL is completed, each vial can be gently swirled and/or inverted slowly for at least 2 minutes until complete dissolution of any cake/powder occurs. Generation of foam should be avoided.
- the concentration of each vial should be 100 mg/10 mL ABX and 40 mg/10 mL BEV.
- the vials containing the ABX and BEV should sit for 60 minutes.
- the vial(s) should be gently swirled and/or inverted every 10 minutes to continue to mix the complex.
- the calculated dosing volume of ABX and BEV should be withdrawn from each vial and slowly added to an empty viaflex bag.
- An equal volume of 0.9% Sodium Chloride Injection, USP is then added to make the final concentration of ABX 5 mg/mL and BEV 2 mg/mL.
- the bag should then be gently swirled and/or inverted slowly for 1 minute to mix.
- the ABX:BEV nanoparticles can be stored for up to 4 hours at room temperature following final dilution.
- Example 1 To determine whether ABX and BEV interact, the nanoparticles formed in Example 1 were analyzed by flow cytometry and electron microscopy.
- AB160 was produced as described in Example 1 above. To determine binding of BEV to ABX, visualization of AB 160 was performed on an Accuri C6 flow cytometer (BD Franklin Lakes, NJ) and data analysis was done using Accuri C6 software. Biotinylated ⁇ g) goat anti-mouse IgG (Abeam, Cambridge, MA) was labeled with 5 ⁇ g of streptavidin PE (Abeam, Cambridge, MA). The goat anti-mouse IgG was chosen to label AB160 because the Fab portion of the BEV is mouse derived.
- ABX and AB160 were incubated with the PE-labeled goat anti-mouse IgG for 30 minutes at room temperature, washed and visualized by flow cytometry.
- Electron Microscopy Five ⁇ ABX, dissolved in PBS at 6 mg/ml, was added to a 300-mesh parlodian-carbon coated copper grid and allowed to sit for 1 minute. A pointed piece of filter paper was touched to the drop to remove excess liquid, leaving a thin film on the grid. The grids were allowed to dry. To dissolve the buffer crystals left on the dried grid, the sample was washed three times in dH 2 0. A small drop of 1% phosphotungstic acid (PTA), pH 7.2, was added to the grid.
- PTA 1% phosphotungstic acid
- BEV (Genentech) at 25 mg/ml in 0.9% sodium chloride solution was diluted with PBS at 1 : 10 ratio. Five ⁇ of BEV was loaded on nickel formvar-coated grid and allowed to air dry for 30 minutes to 1 hour.
- AB160 10 mg/ml ABX, dissolved in PBS, and 4mg/ml BEV, in 0.9% sodium chloride solution, were mixed at 2.5: 1 ratio.
- the complex was further diluted with PBS at 1 :5. Five ⁇ of the complex was loaded on nickel formvar-coated grid and air dried for 30 minutes to 1 hour.
- ABX (10 mg/ml) was co-incubated with 4 mg/ml BEV in vitro and found that they formed 1 60 nm nanoparticles (referred to herein as AB 160). Because the Fab portion of the IgGI (BEV) is of mouse origin, particles containing BEV were selectively labeled with purified goat anti-mouse IgG followed by anti-goat PE as a secondary antibody. As a negative control, samples were stained with the anti-goat PE only.
- AB 160 were made and collected fractions: the particulate (nanoAB160), proteins greater than 100 kD and proteins less than 100 kD.
- Paclitaxel was measured in each fraction by liquid chromatography-mass spectrometry (LC-MS). Roughly 75% of the paclitaxel remained within the particulate, and the majority of the remaining paclitaxel was associated with the fraction containing proteins 100 kD or greater (FIG. 1 C, top), suggesting that when the particulate dissociates the paclitaxel is bound to BEV alone or a BEV and albumin heterodimer.
- LC-MS liquid chromatography-mass spectrometry
- VEGF ELISA To determine whether BEV can still bind its ligand, VEGF, when bound to ABX, a standard VEGF ELISA (R and D Systems, Minneapolis, MN) was employed. AB 160 was prepared as described and 2000 pg/ml VEGF was added to the AB160 complex or ABX alone. The VEGF was incubated with the nanoparticles for 2 hours at room temperature. The suspension was spun at 6000 rpm for 15 minutes, supernatants were collected and free VEGF was measured by ELISA.
- ELISA plates were coated with capture antibody overnight at 4 °C. Plates were washed, blocked and standards and samples were added. After washing, detection antibody was added and plates were developed with substrate (R and D Systems, Minneapolis, MN). Absorbance was measured at 450 nm using a Versamax ELISA plate reader (Molecular Devices, Sunnyvale, CA). The concentration of unbound VEGF was determined with a standard curve from 0 to 2000 pg/ml.
- AB160 has similar toxicity relative to ABX alone in an in vitro toxicity assay with the human melanoma cell line, A375, suggesting that the paclitaxel functions equally in either formulation (FIG. 1 D).
- AB 160 or ABX was co- incubated with VEGF, the particulate removed, and the supernatant tested for VEGF content.
- the lack of VEGF in the supernatant measured from AB160 ( ⁇ 10% VEGF unbound) indicated that the VEGF was bound by the BEV in the AB 160 complex, while it remained free when incubated with the ABX (>80% VEGF unbound) alone (FIG. 1 E).
- Mastersizer and Nanosight The particle size of ABX and antibody-ABX drug complexes were measured by dynamic light scattering on a Mastersizer 2000 (Malvern Instruments, Westborough, MA). To measure particle size, 2 ml (5 mg/ml) of ABRAXA E® or complex was added to the sample chamber. Data were analyzed with Malvern software and particle size distributions were displayed by volume. The particle sizes and stability were later validated using the Nanosight System (Malvern Instruments, Westborough, MA). The ABX or complex particles were diluted to the appropriate range to accurately measure particle sizes. Data was displayed by particle size distribution;
- nanoparticle tracking analysis uses Brownian motion to determine particle size.
- Binding Assay Biotinylated BEV, rituximab or trastuzumab at 100 ⁇ g/ml was bound to the streptavidin probe (ForteBio Corp. MenloPark, CA). The binding of ABX was measured by light absorbance on the BLitz system (ForteBio Corp. MenloPark, CA) at 1000, 500 and 100 mg/ml. The association and dissociation constants were calculated using the BLitz software.
- Bio-Layer Interferometry (BLitz) technology was utilized to assess the binding affinity of BEV to ABX.
- Biotinylated BEV was bound to the streptavidin probe and exposed to ABX (1000, 500, and 100 ⁇ g/ml).
- ABX is 2.2 x 10 " M at room temperature and pH 7, consistent with a strong non-covalent interaction.
- the binding affinity of BEV and ABX is within the range of dissociation constants observed between albumin and natural or engineered albumin-binding domains of some bacterial proteins. Nilvebrant, J. et al. (2013) Comput Struct Biotechnol J
- ABX:BEV nanoparticles were consistently larger (approximately 160 nm) than the 130 nm ABX alone (Fig 2 A).
- the goal of these studies being a Phase I clinical trial the smallest ABX:BEV particle (AB160) were focused on because it is the most similar to the 130 nm ABX.
- the size of the AB160 particle was consistent with ABX plus a monolayer of BEV surrounding it and with the EM image of the particle (see FIG. IB).
- ABX or AB160 was incubated in saline or heparinized human plasma at relative volume ratios of 9: 1 or 1 : 1. Notably, no particles (0.01 to 1 ⁇ detected when either ABX (FIG. 10, top panel) or AB 160 (FIG. 10, bottom panel) is incubated in plasma at equal volumes (1 : 1).
- the ABX was suspended in BEV and the mixture diluted with saline at pH 3, 5, 7, or 9 prior to incubation at various temperatures (RT, 37 °C and 58 °C) to allow particle formation in order to test whether binding affinity was pH- and/or temperature- dependent.
- the binding affinity of ABX and BEV is both pH- and temperature- dependent, with the highest binding affinity observed when the particles are formed at pH 5 and 58 °C (FIG. 2B).
- AB1600758 was compared to ABX exposed to the same conditions (ABX0558, ABX07, and ABX0758, respectively) after incubation in human AB serum for 0, 15, 30, or 60 minutes.
- the particles made under higher affinity conditions were also more stable, as indicated by the number of particles present per mg ABX after exposure to human AB serum.
- the AB160 particles exhibited increased stability in human serum that correlated with their binding affinities.
- AB16007 and AB 1600558 were more stable in both saline and human serum than ABX alone, as determined by size and number of particles measured per mg ABX (FIG. 2C and Table 3). This shows that the stability of AB160 particles can be manipulated by changing the conditions under which the AB 160 particles are formed.
- a xenograft model of A375 human melanoma cells implanted into athymic nude mice was employed to test AB160 efficacy in vivo.
- BEV 45 mg/kg
- Particles of increasing size were prepared using increasing BEV:ABX ratios as shown in FIG. 2A. Tumor regression and median survival positively correlated with increasing particle size, indicating that biodistribution of larger particles may be altered relative to the smaller ones (FIGs. 3D and 3E). Full toxicity studies were performed on the mice and no toxicities were noted.
- Paclitaxel Pharmacokinetics The liquid chromatographic separation of paclitaxel and d5 paclitaxel were accomplished using an Agilent Poroshell 120 EC-C18 precolumn (2.1 x 5 mm, 2.7 ⁇ , Chrom Tech, Apple Valley, MN) attached to an Agilent Poroshell 120 EC-C18 analytical column (2.1 x 100 mm, 2.7 ⁇ Chrom Tech, Apple Valley, MN) at 40 °C, eluted with a gradient mobile phase composed of water with 0.1% formic acid (A) and ACN with 0.1% formic acid (B) with a constant flow rate of 0.5 ml/minute.
- Agilent Poroshell 120 EC-C18 precolumn 2.1 x 5 mm, 2.7 ⁇ , Chrom Tech, Apple Valley, MN
- Agilent Poroshell 120 EC-C18 analytical column 2.1 x 100 mm, 2.7 ⁇ Chrom Tech, Apple Valley, MN
- paclitaxel and the internal standard d5-paclitaxel were accomplished using the mass spectrometer in positive ESI mode with capillary voltage 1.75 kV, source temp 150 °C, desolvation temp 500 °C, cone gas flow 150 L/hr, desolvation gas flow 1000 L/hr, using multiple reaction monitoring (MRM) scan mode with a dwell time of 0.075 seconds.
- MRM multiple reaction monitoring
- the cone voltages and collision energies were determined by MassLynx-Intelli start, v4.1, software and varied between 6-16 V and 12-60 eV, respectively.
- the MRM precursor and product ions were monitored at m/z 854.3> 105.2 for paclitaxel and 859.3>291.2 for d5 paclitaxel.
- the primary stock solutions of paclitaxel (1 mg/ml in EtOH) and d5 paclitaxel (1 mg/ml in EtOH) were prepared in 4 ml amber silanized glass vials and stored at -20 °C.
- Working standards were prepared by dilution of the stock solution with ACN in 2 ml amber silanized glass vials and stored at -20 °C.
- Plasma samples were extracted as follows, 100 ⁇ plasma sample was added to a 1.7 ml microcentrifuge tube containing d5 paclitaxel (116.4 nM or 100 ng/ml) and 300 ⁇ ACN, vortexed, incubated at room temperature for 10 minutes to precipitate proteins, and centrifuged (14,000 rpm) or 3 minutes. The supernatant was filtered on an Agilent Captiva ND lipids plate (Chrom Tech, Apple Valley, MN), collected in a deep 96-well plate, and dried using nitrogen gas. The samples were reconstituted using 100 ⁇ ACN and shaken on a plate shaker (high speed) for 5 minutes.
- Plasma standard curves were prepared daily containing paclitaxel (0.59-5855 nM or 0.5-5000 ng/ml) and d5 paclitaxel (116.4 nM) for paclitaxel quantitation.
- Mouse tumors were thawed on ice, weighed, and diluted 2 parts (weight to volume) in l x PBS. Tumors were then homogenized using a PRO200 tissue homogenizer using the saw tooth probe (5 mm x 75 mm). Tumor homogenate was than processed the same as the human plasma samples.
- ABRAXA E®-human IgG 1-125, or ABRAXANE® only Animals were imaged at 3, 10, 24 and 72 hours post-administration via SPECT- CT imaging using the U-SPECT- II/CT scanner (MILabs, Utrecht, The Netherlands). SPECT reconstruction was performed using a POSEM (pixelated ordered subsets by expectation maximization) algorithm. CT data were reconstructed during the Feldkamp algorithm. Co-registered images were further rendered and visualized using PMOD software (PMOD Technologies, Zurich, Switzerland). Animals were sacrificed and dissected at 72 hours post- injection. Selected tissues and organs of interest were measured using radioisotope dose calibrator (Capintec CRC-127R, Capintec Inc.).
- Results of the first pk experiment are provided in FIGs. 4A and 4B.
- the area under the curve (AUC) and maximum serum concentration (Cmax) were calculated in A375 tumor bearing and non-tumor bearing mice.
- the Cmax and AUC were very similar in the non-tumor bearing mice for AB160 and ABX (63.3+ ⁇ 39.4 vs. 65.5+/-14.4 and 129 vs. 133 ⁇ g/ml, respectively).
- the Cmax and AUC for the treatment groups were different (55.7+/-21.2 vs 63.3+/- 17.3 and 112 vs 128 ⁇ g/ml, respectively) (FIG. 4C). Although this difference was not statistically significant, it is consistent with superior targeting by AB 160, relative to ABX.
- FIGs. 4D-4F A second pk experiment was performed with additional early time points and large versus small tumor sizes (FIGs. 4D-4F). The results of this experiment demonstrated smaller AUC in tumor bearing mice relative to non-tumor bearing mice, with the lowest blood values of paclitaxel in the large tumor mice relative to the small tumor mice (80.4+/-2.7, 48.4+/-12.3, and 30.7+/-5.2 for ABX-treated non-tumor, small tumor and large tumor bearing mice, respectively; 66.1+/-19.8, 44.4+/-12.1 and 22.8+/-6.9 for AB160-treated).
- tumor paclitaxel concentrations by LC-MS were measured.
- mice blood paclitaxel pk, tumor tissue levels of paclitaxel, and 1-125 radioactivity levels in mice treated with AB160 relative to ABX alone suggest that antibody targeting of the ABX alters biodistribution of paclitaxel such that increased levels reach the tumor and are retained there for a longer period of time, yielding enhanced tumor regression.
- Nanoparticles containing rituximab or trastuzumab were prepared and tested as described in the above examples.
- AR160 160 nm particle made with rituximab
- a xenotransplant model of Daudi cells was established in athymic nude mice. Once tumors were established, mice were treated with PBS, ABX, rituximab, ABX and rituximab given sequentially, or AR160. On day 7 post treatment, tumors were measured and the percent change in tumor size from baseline was calculated. AR160- treated tumors regressed or remained stable, while tumors in all other treatment groups progressed (FIG. 6B). The percent change from baseline tumor size in the AR160 group compared to all other groups was significant (p ⁇ 0.0001).
- Nanoparticles containing cisplatin were prepared and tested as described in the above examples.
- AB 160 was co-incubated with cisplatin to form cisplatin containing particles (ABC complex).
- the ABC complex was tested in the A375 melanoma xenotransplant model relative to each drug alone and AB 160. Tumors treated with AB160, AB160 + cisplatin given
- albumin portion of the ABX provides a platform for other therapeutic antibodies to bind, such as rituximab and trastuzumab, as well as other chemotherapy agents (e.g., cisplatin), which all had similar efficacy in vitro and in vivo as AB160.
- AB160 was synthesized by adding 8mg (320 ⁇ 1) of bevacizumab to 20mg of ABRAXA E®. 1.66ml of 0.9% saline was then added for a final volume of 2ml for a final concentration of 4mg/ml bevacizumab and 1 0 m g/ml ABRAXANE®, and the mixture was allowed to incubate at room temperature for 30 minutes in a 15ml polypropylene conical tube.
- the mixture was diluted 1 :2 in 0.9% saline to 2mg/ml and 5mg/ml bevacizumab and ABRAXA E®, respectively. These are the concentrations of the 2 drugs when prepared by the pharmacy for administration to patients.
- AB160 was divided into twenty 200 ⁇ 1 aliquots in 1.5 ml polypropylene eppendorfs and frozen at -80 °C.
- polypropylene eppendorfs These samples were readily reconstituted in saline at room temperature for 30 minutes, followed by centrifugation for 7 minutes at 2000x g. The resulting sample was then resuspended in the appropriate buffer, as needed.
- VEGF binding was evaluated by incubation of the sample with VEGF for 2 hours at room temperature, centrifuged at 2000 x g for 7 minutes. The amount of VEGF bound to the pellet (corresponding to the nanoparticles) or remaining in the supernatant was measured with ELISA.
- Paclitaxel activity was assessed by cytotoxicity against A375 cells in vitro.
- lyophilization did not significantly affect either the particle size, VEGF binding, or the activity of paclitaxel as shown by the ability to inhibit cancer cell proliferation. This result held for lyophilized samples stored for 1 month (FIGs. 8A-8C) or 10 months (FIGs. 8D-8F).
- AB160 was tested in a phase 1, first-in-man, clinical trial testing the safety of AB160 administered to patients with metastatic malignant melanoma that have failed prior therapies.
- the study utilizes a classical 3+3, phase 1 clinical trial design, testing 3 different doses of AB160 in the following schema:
- Dose level 1 refers to the starting dose.
- the doses were selected relevant to doses of ABRAXA E® currently used in clinical practice. AB 160 was made prior to each treatment dose. Treatments were administered as a 30 minute intravenous infusion on days 1, 8 and 15 of a 28-day treatment cycle. Treatments were continued until intolerable toxicity, tumor progression or patient refusal. Prior to every treatment cycle, patients were evaluated for toxicity; tumor evaluations were performed every other cycle (RECIST).
- PFS refers to median progression free survival, i.e. the number of days of treatment before the cancer recurred. Adverse events are listed below. There was no dose limiting toxicity (DLT), i.e. the adverse events were not linked to the dose of AB 160. More detail is provided in Table 6.
- DLT dose limiting toxicity
- the ABX/BEV complex (AB160) is superior to sequential administration of ABX and BEV, or ABX alone, and achieves this superior result with a very low effective dose of BEV.
- the data is therefore consistent with AB160 having improved targeting of the chemotherapeutics to the tumor, and that this targeting is mediated by BEV.
- the ABX nanoparticle aids in targeting the BEV to the tumor, as albumin is selectively taken up by tumors.
- the existence of the BE ABX complex shows greater stability in vivo than ABRAXANE®.
- Athymic nude mice were injected with 1 x 10 A375 human melanoma cells in the right flank and then treated with PBS, 12 mg/kg BEV, 30 mg/kg ABX, AB160, or pretreated with 1.2 mg/kg BEV and, 24hr later, AB 160.
- PBS 12 mg/kg BEV
- ABX 30 mg/kg ABX
- AB160 pretreated with 1.2 mg/kg BEV and, 24hr later, AB 160.
- mice represented at day 7-post and day 10 -post treatment as tumor volume in mm .
- F 1 1A-E track tumor size over 10 days. Only mice treated with AB160 (with or without
- pretreatment with BEV showed a reduction in average tumor volume. See also FIG. 1 1 F and FIG. 1 1 G.
- Example 13 Alternative means of delivering nanoparticles
- nanoparticles of this invention can be directly delivered to the tumor.
- nanoparticles can be delivered via intra-arterial cannula or by direct injection into the tumor.
- large nanoparticles e.g., 580 nm or 1130 nm
- nanoparticles can be delivered by direct injection into or proximate to a tumor.
- CD20 positive Daudi lymphoma cells were labeled with fluorescent tagged anti- human CD20 or isotype matched control in panel F and A, respectively, and analyzed by flow cytometry.
- the Daudi cells were pretreated with ABX, AR160, AR160L (AR160 lyophilized and resuspended into a solution suitable for injection), or Rituxan prior to CD20 labeling.
- Figure 12 demonstrates that CD20 binding was specifically blocked by the AR particles and Rituxan, but not ABX alone.
- Figure 13 is a histogram overlay of the same data presented in Figure 12.
- FIG 14A and 14B depicts the particle size comparisons of ABX alone relative to AR (FIG. 14A) and AT (FIG. 14B) freshly made and lyophilized.
- FIG 15 presents the results of a Daudi proliferation assay comparing the toxicity of ABX and the AR particles. The data demonstrates the lyophilized and non-lyophilized nanoparticles have essentially the same toxicity in the Daudi assay.
- Example 15 Fluorescent analysis of tumor accumulation of AlexaFluor 750 labeled
- FIG. 16A is a bar graph of the average fluorescence per unit of tumor area of mice in all three treatment groups were determined to provide the gross tumor delivery.
- FIG. 16 C is a bar graph of the average fluorescence per unit of tumor area normalized by background ROI to give proportion of drug delivered to tumor versus body.
- AR160 nanoparticles results in an increased fluorescence as compared to ABRAXANE® alone or ABRAXANE® coated with non-specific antibodies.
- Example 16 Nanoparticles having a size of 225nm
- FIG. 17 depicts the survival of the mice treated with a single dose of saline, BEV, ABX, AB 160 and AB225 and with AB160 with a BEV pretreatment. At 30 days post-administration the survival of mice treated with AB225, and with AB160 with or without pretreatment with BEV far exceeds the survival of mice treated with BEV alone of ABRAXANE® alone.
Abstract
Described herein are compositions of antibodies and carrier proteins and methods of making and using the same, in particular, as a cancer therapeutic. Also described are lyophilized compositions of antibodies and carrier proteins and methods of making and using the same, in particular, as a cancer therapeutic.
Description
CARRIER-BINDING AGENT COMPOSITIONS AND METHODS OF MAKING AND
USING THE SAME
RELATED APPLICATIONS
[0001] The disclosures of PCT Application No. PCT/US 15/54295, filed October 6, 2015, U.S. Provisional Patent Application No. 62/060,484, filed October 6, 2014, U.S. Provisional Patent Application No. 62/206,770; 62/206,771 filed August 18, 2015, and U.S. Provisional Patent Application No.62/206,772 filed August 18, 2015 are incorporated by reference in their entireties.
FIELD OF THE INVENTION
[0002] This application relates to novel compositions of binding agents and carrier proteins and methods of making and using the same, in particular, as a cancer therapeutic.
STATE OF THE ART
[0003] Chemotherapy remains a mainstay for systemic therapy for many types of cancer, including melanoma. Most chemotherapeutics are only slightly selective to tumor cells, and toxicity to healthy proliferating cells can be high (Allen TM. (2002) Cancer 2:750-763), often requiring dose reduction and even discontinuation of treatment. In theory, one way to overcome chemotherapy toxicity issues as well as improve drug efficacy is to target the chemotherapy drug to the tumor using antibodies that are specific for proteins selectively expressed (or overexpressed) by tumors cells to attract targeted drugs to the tumor, thereby altering the biodistribution of the chemotherapy and resulting in more drug going to the tumor and less affecting healthy tissue. Despite 30 years of research, however, specific targeting rarely succeeds in the therapeutic context.
[0004] Conventional antibody dependent chemotherapy (ADC) is designed with a toxic agent linked to a targeting antibody via a synthetic protease-cleavable linker. The efficacy of such ADC therapy is dependent on the ability of the target cell to bind to the antibody, the linker to be cleaved, and the uptake of the toxic agent into the target cell. Schrama, D. et al. (2006) Nature reviews. Drug discovery 5: 147-159.
[0005] Antibody-targeted chemotherapy promised advantages over conventional therapy because it provides combinations of targeting ability, multiple cytotoxic agents, and improved therapeutic capacity with potentially less toxicity. Despite extensive research,
clinically effective antibody -targeted chemotherapy remains elusive: major hurdles include the instability of the linkers between the antibody and chemotherapy drug, reduced tumor toxicity of the chemotherapeutic agent when bound to the antibody, and the inability of the conjugate to bind and enter tumor cells. In addition, these therapies did not allow for control over the size of the antibody-drug conjugates.
[0006] There remains a need in the art for antibody -based cancer therapeutics that retain cytotoxic effect for targeted drug delivery to provide reliable and improved anti-tumor efficacy over prior therapeutics.
[0007] In addition, as to any therapeutic application, there also remains a need for the composition to be stable in its physical, chemical and biological properties.
[0008] Lyophilization, or freeze-drying, removes water from a composition. In the process, the material to be dried is first frozen and then the ice or frozen solvent is removed by sublimation in a vacuum environment. An excipient may be included in pre-lyophilized formulations to enhance stability during the freeze-drying process and/or to improve stability of the lyophilized product upon storage. Pikal, M. Biopharm. 3(9)26-30 (1990) and Arakawa et al., Pharm. Res. 8(3):285- 291 (1991).
[0009] While proteins may be lyophilized, the process of lyophilization and reconstitution may affect the properties of the protein. Because proteins are larger and more complex than traditional organic and inorganic drugs (i.e. possessing multiple functional groups in addition to complex three-dimensional structures), the formulation of such proteins poses special problems. For a protein to remain biologically active, a formulation must preserve intact the conformational integrity of at least a core sequence of the protein's amino acids while at the same time protecting the protein's multiple functional groups from degradation. Degradation pathways for proteins can involve chemical instability (i.e. any process which involves modification of the protein by bond formation or cleavage resulting in a new chemical entity) or physical instability (i.e. changes in the higher order structure of the protein). Chemical instability can result from deamidation, racemization, hydrolysis, oxidation, beta elimination or disulfide exchange. Physical instability can result from denaturation. aggregation, precipitation or adsorption, for example. The three most common protein degradation pathways are protein aggregation, deamidation and oxidation. Cleland, et al., Critical Reviews in Therapeutic Drug Carrier Systems 10(4): 307-377 (1993).
SUMMARY
[0010] In the present invention, the composition comprises nanoparticles which contain (a) carrier protein (b) a binding agent, and (c) optionally a therapeutic agent. The binding agent is believed to be bound to the carrier protein through hydrophobic interactions which, by their nature, are weak. Yet the activity of the individual components, as well as their relative relationship in the nanoparticle are preserved despite lyophilization and reconstitution of the a composition. It is still further contemplated that binding to the carrier protein, e.g., complexation of the binding agent to the carrier protein, occurs through some or all of the hydrophobic portion of the binding agent, e.g., the Fc component, which results in all or part of the hydrophobic portion being integrated into the carrier protein core, while the target binding portions (regions) (e.g., an Fa and Fb portion) of the antibody remain outside of the carrier protein core, thereby retaining their target specific binding capabilities. In some embodiments, the binding agent is a non-therapeutic and non-endogenous human antibody, a fusion protein, e.g., fusion of an antibody Fc domain to a peptide that binds a target antigen, or an aptamer.
[0011] Further challenges are imposed because the nanoparticles are used in therapy.
[0012] While rearrangement of the hydrophobic components in the nanoparticle may be mitigated through covalent bonds between the components, such covalent bonds pose challenges for the therapeutic use of nanoparticles in cancer treatment. The binding agent, carrier protein, and additional therapeutic agent typically act at different locations in a tumor and through different mechanisms. Non-covalent bonds permit the components of the nanoparticle to dissociate at the tumor. Thus, while a covalent bond may be advantageous for lyophilization, it may be disadvantageous for therapeutic use.
[0013] The size of nanoparticles, and the distribution of the size, is also important.
Nanoparticles may behave differently according to their size. At large sizes, nanoparticles or the agglomeration of the particles may block blood vessels either of which can affect the performance and safety of the composition.
[0014] Finally, cryoprotectants and agents that assist in the lyophilization process must be safe and tolerated for therapeutic use.
[0015] In the present invention, the inventive compositions comprise nanoparticles which nanoparticles contain (a) carrier protein (b) a binding agent and (c) optionally a therapeutic agent. Without wishing to be bound by theory, the binding agent is believed to be bound to the carrier protein through hydrophobic interactions which, by their nature, are weak. Yet, the activity of the individual components, and their relative relationship in the nanoparticle are still achieved despite lyophilization and reconstitution of the composition.
[0016] In one aspect, provided herein are nanoparticle compositions comprising
nanoparticles wherein each of the nanoparticles comprises a carrier protein, between about 100 to about 1000 binding agents, and optionally at least one therapeutic agent, wherein the binding agents are arranged outward from the surface of the nanoparticles and wherein the nanoparticles are capable of binding to a predetermined epitope in vivo.
[0017] When administered intravenously, large particles (e.g. greater than 1 μιη) are typically disfavored because they can become lodged in the microvasculature of the lungs. At the same time, larger particles can accumulate in the tumor or specific organs. See e.g. 20-60 micron glass particle that is used to inject into the hepatic artery feeding a tumor of the liver, called "TheraSphere" (in clinical use for liver cancer).
[0018] Therefore, for intravenous administration, particles under 1 μπι are used. Particles over 1 μπι are, more typically, administered directly into a tumor ("direct injection") or into an artery feeding into the site of the tumor.
[0019] In another aspect, provided herein are nanoparticle compositions comprising nanoparticles wherein each of the nanoparticles comprises a carrier protein that is not albumin, between about 100 to about 1000 binding agents, preferably about 400 to about 800 binding agents, and optionally at least one therapeutic agent, wherein the binding agents are arranged on an outside surface of the nanoparticles and wherein the nanoparticles are capable of binding to a predetermined epitope in vivo. When nanoparticles multimerize, the number of binding agents is increased proportionally. For example, if a 160 nm nanoparticle contains 400 binding agents, a 320 nm dimer contains about 800 binding agents.
[0020] In another aspect, provided herein are nanoparticle compositions comprising nanoparticles, wherein each of the nanoparticles comprises carrier protein, between about 400 to about 800 binding agents, and optionally at least one therapeutic agent that is not paclitaxel, wherein the binding agents are arranged on a surface of the nanoparticles such that
the binding portion of the binding agent is directed outward from that surface and wherein the nanoparticles are capable of binding to a predetermined epitope in vivo.
[0021] In other embodiments, the nanoparticles multimerize, e.g. dimerize. Multimerization may be observed as multiples of the weight or size of the unit molecule, e.g. 160 nm particles multimerize to about 320 nm, 480 nm, 640 nm, etc. In some embodiments, less than 20% of the nanoparticles in a population are multimers. In some embodiments, more than 80% of the nanoparticles in a population are multimers.
[0022] In one embodiment, the weight ratio of carrier-bound drug to binding agent (e.g. albumin- bound paclitaxel to bevacizumab) is between about 5: 1 to about 1 : 1. In one embodiment, the weight ratio of carrier-bound drug to binding agent is about 10:4. In one embodiment, the binding agents are a substantially single layer on all or part of the surface of the nanoparticle. In one embodiment, less than 0.01% of nanoparticles in the composition have a size selected from the group consisting of greater than 200 nm, greater than 300 nm, greater than 400 nm, greater than 500 nm, greater than 600 nm, greater than 700 nm and greater than 800 nm. Larger sizes are believed to be the result of multimerization of several nanoparticles, each comprising a core and binding agent coating on all or part of the surface of each nanoparticle.
[0023] The invention further includes lyophilized compositions, and lyophilized
compositions that do not materially differ from, or are the same as, the properties of freshly- prepared nanoparticles. In particular, the lypholized composition, upon resuspending in aqueous solution, is similar or identical to the fresh composition in terms of particle size, particle size distribution, toxicity for cancer cells, binding agent affinity, and binding agent specificity. The invention is directed to the surprising finding that lyophilized nanoparticles retain the properties of freshly-made nanoparticles after resuspension notwithstanding the presence of two different protein components in these particles.
[0024] In one aspect, this invention relates to a lyophilized nanoparticle composition comprising nanoparticles, wherein each of the nanoparticles comprises a carrier-bound drug core and an amount of binding agent arranged on a surface of the core such that the binding portion of the binding agent is directed outward from that surface, wherein the binding agents retain their association with the outside surface of the nanoparticle upon reconstitution with an aqueous solution. In one embodiment, the lyophilized composition is stable at room
temperature for at least 3 months. In one embodiment, the reconstituted nanoparticles retain the activity of the therapeutic agent and are capable of binding to the target in vivo.
[0025] In one embodiment, the average reconstituted nanoparticle size is from about 130 nm to about 1 μιη. In a preferred embodiment, the average reconstituted nanoparticle size is from about 130 nm to about 200 nm, and more preferably about 160 nm. In one embodiment, in the average reconstituted nanoparticle size is from greater than 800 nm to about 3.5 μιτι, comprising multimers of smaller nanoparticles, e.g. multimers of 100-200 nm nanoparticles. In one embodiment, the weight ratio of core to binding agent is from greater than 1 : 1 to about 1 :3. In one embodiment, in the average reconstituted nanoparticle size is about 160nm to about 225nm.
[0026] In one aspect, this disclosure relates to a lyophilized nanoparticle composition comprising nanoparticles, wherein each of the nanoparticles comprises: (a) an albumin-bound paclitaxel core and (b) between about 400 to about 800 molecules of bevacizumab arranged on a surface of the albumin-bound paclitaxel core such that the binding portion of the binding agent is directed outward from that surface, wherein the binding agents retain their association with the surface of the nanoparticle upon reconstitution with an aqueous solution, provided that said lyophilized composition is stable at about 20 °C to about 25 °C for at least 3 months and the reconstituted nanoparticles are capable of binding to VEGF in vivo.
[0027] In other aspects, this disclosure relates to a lyophilized nanoparticle composition comprising nanoparticles, wherein each of the nanoparticles comprises: (a) an albumin-bound paclitaxel core and (b) an amount of bevacizumab arranged on a surface of the albumin- bound paclitaxel core such that the binding portion of the binding agent is directed outward from that surface, wherein the binding agents retain their association with the surface of the nanoparticle upon reconstitution with an aqueous solution, provided that said lyophilized composition is stable at about 20 °C to about 25 °C for at least 3 months and the reconstituted nanoparticles are capable of binding to VEGF in vivo, and further wherein the average reconstituted nanoparticle size is not substantially different from the particle size of the freshly prepared nanoparticles. In some embodiments, the average particle sizes are between 200 and 800 nm, including 200, 300, 400, 500, 600, 700 or 800nm. In other embodiments, the average particles are larger, e.g. from greater than 800 nm to about 3.5 μιη. In some embodiments, the particles are multimers of nanoparticles. In some embodiments the
nanoparticles have average particle sizes of about 160nm to about 225 nm either freshly made or after lyophilization and resuspension in an aqueous solution suitable for injection.
[0028] In some embodiments, the weight ratio of albumin-bound paclitaxel to bevacizumab is between about 5: 1 to about 1 : 1. In other embodiments, the weight ratio of albumin-bound paclitaxel to bevacizumab is about 10:4. In further embodiments, the weight ratio of albumin- bound paclitaxel to bevacizumab is from greater than 1 : 1 to about 1 :3.
[0029] In some embodiments, the core is albumin-bound paclitaxel, and the binding agents are selected from binding agents that selectively recognize VEGF (e.g. bevacizumab/ Avastin), binding agents that selectively recognize CD20 (e.g. rituximab/Rituxin) and binding agents that selectively recognize Her2 (Trastuzumab/Herceptin).
[0030] In some embodiments, the at least one therapeutic agent is located inside the nanoparticle. In other embodiments, the at least one therapeutic agent is located on the outside surface of the nanoparticle. In yet other embodiments, the at least one therapeutic agent is located inside the nanoparticle and on the outside surface of the nanoparticle.
[0031] In some embodiments, the nanoparticle contains more than one type of therapeutic agent. For example, a taxane and a platinum drug, e.g. paclitaxel and cisplatin.
[0032] In some embodiments, the binding agents are selected from the group consisting of ado- trastuzumab emtansine, alemtuzumab, bevacizumab, cetuximab, denosumab, dinutuximab, ipilimumab, nivolumab, obinutuzumab, ofatumumab, panitumumab, pembrolizumab, pertuzumab, rituximab, and trastuzumab. In some embodiments, the binding agents are a substantially single layer of binding agents on all or part of the surface of the nanoparticle.
[0033] In further embodiments, the antibodies are less glycosylated than normally found in natural human antibodies. Such glycosylation can be influenced by e.g. the expression system, or the presence of glycosylation inhibitors during expression. In some embodiments, the glycosylation status of an antibody or other binding agent is altered through enzymatic or chemical action.
[0034] In some embodiments, the at least one therapeutic agent is selected from the group consisting of abiraterone, bendamustine, bortezomib, carboplatin, cabazitaxel, cisplatin,
chlorambucil, dasatinib, docetaxel, doxorubicin, epirubicin, erlotinib, etoposide, everolimus, gefitinib, idarubicin, imatinib, hydroxyurea, imatinib, lapatinib, leuprorelin, melphalan, methotrexate, mitoxantrone, nedaplatin, nilotinib, oxaliplatin, paclitaxel, pazopanib, pemetrexed, picoplatin, romidepsin, satraplatin, sorafenib, vemurafenib, sunitinib, teniposide, triplatin, vinblastine, vinorelbine, vincristine, and cyclophosphamide.
[0035] In some embodiments, the nanoparticle further comprises at least one additional therapeutic agent that is not paclitaxel or bevacizumab.
[0036] In some embodiments, the binding agents, carrier protein and, when present, therapeutic agent, are bound through non-covalent bonds.
[0037] In some embodiments, the carrier protein is selected from the group consisting of gelatin, elastin, gliadin, legumin, zein, a soy protein, a milk protein, and a whey protein. In other embodiments, the carrier protein is albumin, for example, human serum albumin.
[0038] In some embodiments, the composition is formulated for intravenous delivery. In other embodiments, the composition is formulated for direct injection or perfusion into a tumor.
[0039] In some embodiments, the average nanoparticle size in the composition is from greater than 800 nm to about 3.5 μιη.
[0040] In some embodiments, the nanoparticles have a dissociation constant between about 1 x 10"11 M and about 1 x 10"9 M.
[0041] In another aspect, provided herein are methods of making nanoparticle compositions, wherein said methods comprise contacting the carrier protein and the optionally at least one therapeutic agent with the antibodies in a solution having a pH of between 5.0 and 7.5 and a temperature between about 5°C and about 60°C, between about 23°C and about 60°C, or between about 55°C and about 60°C under conditions and ratios of components that will allow for formation of the desired nanoparticles. In one embodiment, the nanoparticle is made at 55- 600C and pH 7.0. In another aspect, provided herein are methods of making the nanoparticle compositions, wherein said method comprises (a) contacting the carrier protein and optionally the at least one therapeutic agent to form a core and (b) contacting the core with the antibodies in a solution having a pH of about 5.0 to about 7.5 at a temperature
between about 5°C and about 60°C, between about 23°C and about 60°C, or between about 55°C and about 60°C under conditions and ratios of components that will allow for formation of the desired nanoparticles.
[0042] The amount of components (e.g., carrier protein, antibodies, therapeutic agents, combinations thereof) is controlled in order to provide for formation of the desired nanoparticles. A composition wherein the amount of components is too dilute will not form the nanoparticles as described herein. In a preferred embodiment, weight ratio of carrier protein to binding agent is 10:4. In some embodiments, the amount of carrier protein is between about 1 mg/mL and about 100 mg/mL. In some embodiments, the amount of binding agent is between about 1 mg/mL and about 30 mg/mL. For example, in some embodiments, the ratio of carrier protein: binding agent: solution is approximately 9 mg of carrier protein (e.g., albumin) to 4 mg of binding agent (e.g., BEV) in 1 mL of solution (e.g., saline). An amount of therapeutic agent (e.g., paclitaxel) can also be added to the carrier protein.
[0043] In further embodiments, the nanoparticles are made as above, and then lyophilized.
[0044] In another aspect, provided herein are methods for treating a cancer cell, the method comprising contacting the cell with an effective amount of a nanoparticle composition disclosed herein to treat the cancer cell.
[0045] In another aspect, provided herein are methods for treating a tumor in a patient in need thereof, the method comprising contacting the cell with an effective amount of a nanoparticle composition disclosed herein to treat the tumor. In some embodiments, the size of the tumor is reduced. In other embodiments, the nanoparticle composition is administered intravenously. In yet other embodiments, the nanoparticle composition is administered by direct injection or perfusion into the tumor.
[0046] In some embodiments, the methods provided herein include the steps of: a) administering the nanoparticle composition once a week for three weeks; b) ceasing administration of the nanoparticle composition for one week; and c) repeating steps a) and b) as necessary to treat the tumor.
[0047] In related embodiments, the treatment comprises administration of the targeting binding agent prior to administration of the nanoparticles. In one embodiment, the targeting
binding agent is administered between about 6 and 48, or 12 and 48 hours prior to
administration of the nanoparticles. In another embodiment, the targeting binding agent is administered between 6 and 12 hours prior to administration of the nanoparticles. In yet another embodiment, the targeting binding agent is administered between 2 and 8 hours prior to administration of the nanoparticles. In still other embodiments, the targeting binding agent is administered a week prior to administration of the nanoparticles. For example,
administration of a dose of BEV 24 hours prior to administration of AB 160. In another example, prior administration of rituximab prior to administering AR nanoparticles. The binding agent administered prior to the nanoparticle may be administered as a dose that is subtherapeutic, such as 1/2, 1/lOth or 1/20 the amount normally considered therapeutic. Thus, in humans, pretreatment with BEV may comprise administration of lmg/kg BEV which is 1/lOth the usual dose, followed by administration of AB160.
[0048] In some embodiments, the therapeutically effective amount comprises about 75 mg/m2 to about 175 mg/m2 of the carrier protein (i.e., milligrams carrier protein per m2 of the patient). In other embodiments, the therapeutically effective amount comprises about 75 mg/m2 to about 175 mg/m2 of therapeutic agent (e.g., paclitaxel). In other embodiments, the therapeutically effective amount comprises about 30 mg/m2 to about 70 mg/m2 of the binding agent. In yet other embodiments, the therapeutically effective amount comprises about 30
2 2
mg/m to about 70 mg/m bevacizumab.
[0049] In one specific embodiment, the lypholized composition comprises from about 75
2 2
mg/m to about 175 mg/m of the carrier protein which is preferably albumin; from about
2 2
30 mg/m to about 70 mg/m of the binding agent which is preferably bevacizumab; and
2 2
from about 75 mg/m to about 175 mg/m of paclitaxel.
[0050] An embodiment of the invention includes a method for increasing the duration of tumor uptake of a chemotherapeutic agent by administering the chemotherapeutic agent in a nanoparticle comprising a carrier protein and the chemotherapeutic agent having surface complexation with an antibody, e.g., an antibody that specifically binds to an antigen on or shed by the tumor.
BRIEF DESCRIPTION OF THE DRAWINGS
[0051] The following figures are representative only of the invention and are not intended as a limitation. For the sake of consistency, the nanoparticles of this invention using ABRAXA E® and bevacizumab employ the acronym "AB" and the number after AB such as AB160 is meant to confer the average particle size of these nanoparticles (in nanometers). Likewise, when the binding agent is rituximab, the acronym is "AR" while the number thereafter remains the same.
[0052] FIG. 1A shows flow cytometry scatterplots including: ABRAXANE® (ABX - commercially available from Celgene Corporation, Summit, NJ 07901) stained with secondary antibody only (top left panel), ABX stained with goat anti-mouse IgGl Fab plus secondary antibody (top right panel), AB160 (which is a nanoparticle of albumin-bound paclitaxel to bevacizumab in a ratio of about 10:4 and has an average particle size of 160 nm) stained with secondary antibody only (bottom left panel), or AB 160 stained with goat anti-mouse IgGl Fab plus secondary antibody (bottom right panel).
[0053] FIG. IB shows a representative electron micrograph after incubation of AB160 with gold particle-labeled anti-human IgG Fe.
[0054] FIG. 1C shows a pie chart (top) indicating the percentages of total paclitaxel in AB 160 fractions (particulate, proteins greater than 100 kD and proteins less than 100 kD); and a Western blot with antibodies against mouse IgG Fab (BEV) and paclitaxel to verify co-localization (bottom).
[0055] FIG. 1 D represents the activity of paclitaxel in an in vitro toxicity assay with A375 human melanoma cells, compared to ABX alone. The results are represented by the average (+/- SEM) proliferation index, which is the percentage of total proliferation of untreated cells. This data represents 3 experiments and differences were not significant.
[0056] FIG. IE represents results from a VEGF ELISA of supernatant after co-incubation of VEGF with ABX and AB160 to determine binding of the ligand, VEGF, by the antibody. The results are shown as the average percentage +/- SEM of VEGF that was unbound by the 2 complexes. The data represents 3 experiments ** P<0.005.
[0057] FIG. 2A shows the size of the complexes (determined by light scattering technology) formed by adding BEV (bevacizumab) to ABX under conditions where nanoparticles and higher are formed. Increasing concentrations of BEV (0-25 mg) were
added to 10 mg of ABX and the size of the complexes formed was determined. The average size of the complexes (146 nm to 2, 166 nm) increased as the concentration of BEV was increased. The data is displayed as volume of sample/size and graphs show the size distribution of the particles. This data is representative of 5 separate drug
preparations. As a comparison, ABX, by itself, has an average particle size of about 130 nm.
[0058] FIG. 2B shows affinity of the binding of ABX and BEV (as determined by light absorption (BLItz) technology). The data is displayed as dissociation constant (Kd). The binding affinity of particles made at four pH levels (3, 5, 7, 9) and 3 temperatures (RT, 37°C and 58°C) was assessed, and the data are representative of 5 experiments.
[0059] FIG. 2C shows the stability of the nanoparticle complexes from Figure 2B in serum as determined by a nanoparticle tracking analysis (NTA) on Nanosight 300
(NS300). The data are displayed as the number of particles/mg of ABX and compares AB160 prepared at RT and pH 7 (AB16007; particle size, pH), 58 °C and pH 7
(AB1600758; particle size, pH, temperature) and 58 °C and pH 5 (AB1600558; particle size, pH, temperature), relative to ABX alone under each condition. Once particles were prepared, they were added to human AB serum for 15, 30, and 60 minutes to determine stability in serum over time.
[0060] FIG. 3A shows in vivo testing of AB nanoparticles in athymic nude mice injected
6
with 1 x 10 A375 human melanoma cells in the right flank and treated with PBS, 12 mg/kg BEV, 30 mg/kg ABX, 12 mg/kg BEV + 30 mg/kg ABX, or AB 160 (having about
3
12 mg/kg BEV and about 30 mg/kg ABX) at tumor size between approximately 600 mm
3
to 900 mm Data is represented at day 7-post treatment as the percent change in tumor size from baseline (the size of the tumor on the day of treatment). Student's t-test was used to determine significance. The p-values for the AB particles were all significantly different than PBS, the individual drugs alone and the 2 drugs given sequentially.
[0061] FIG. 3B shows Kaplan-Meier curves generated for median survival of the mice analyzed in FIG. 3 A. Significance was determined using the Mantle-Cox test comparing survival curves.
[0062] FIG. 3C shows the percent change from baseline for mice treated when tumors
3
were less than or greater than 700 mm , to ascertain whether the size of the tumor affected tumor response for the ABX only and AB 160 groups. The Student's t-test was used to determine significance; the ABX only groups showed no significant difference (p = 0.752) based on tumor size, while the AB 160 groups were significantly different (p = 0.0057).
[0063] FIG. 3D shows in vivo testing of AB nanoparticles in athymic nude mice injected
6
with 1 x 10 A375 human melanoma cells in the right flank and treated with PBS, 30 mg/kg ABX, or 45 mg/kg BEV and AB160, AB580 (nanoparticle of albumin-bound paclitaxel to bevacizumab having an average particle size of 580 nm) or AB1130
(nanoparticle of albumin-bound paclitaxel to bevacizumab having an average particle size
3 3
of 1130 nm) at tumor size between approximately 600 mm to 900 mm . Data is represented at day 7-post treatment as the percent change in tumor size from baseline (the size of the tumor on the day of treatment). Student's t-test was used to determine significance. The changes in tumor size after administration of the AB particles were all significantly different than PBS, the individual drugs alone and the 2 drugs given sequentially. The difference among the AB particles of different sizes was not significant.
[0064] FIG. 3E shows Kaplan-Meier curves generated for median survival of the mice analyzed in FIG. 3D. Significance was determined using the Mantle-Cox test comparing survival curves.
[0065] FIG. 4A demonstrates blood paclitaxel concentration displayed in line graph with y-axis in log scale, based on blood and tumor samples taken from non-tumor and tumor bearing mice at 0-24 hours after IV injection with 30 mg/kg of paclitaxel in the context of ABX or AB 160 and measured by LC-MS. Mice were IV injected at time 0, with blood samples taken and the mice sacrificed at time points of 0, 4, 8, 12, and 24 hours. There were at least 3 mice per time point. Student's t-test was utilized to determine if any differences in concentrations between ABX and AB 160 were significant.
[0066] FIG. 4B demonstrates the blood paclitaxel concentration from FIG. 4A, displayed in line graph with y-axis in numeric scale.
[0067] FIG. 4C shows the Cmax, half-life and AUC values calculated from the blood concentration data provide in FIGs 4 A and 4B.
[0068] FIG. 4D demonstrates blood paclitaxel concentration displayed in line graph with y-axis in log scale from a second pharmacokinetic experiment using earlier time points (2 to 8 hours).
[0069] FIG. 4E demonstrates the blood paclitaxel concentration from FIG. 4D, displayed in line graph with y-axis in numeric scale.
[0070] FIG. 4F shows blood paclitaxel concentration in mice in which the tumors were allowed to grow to a larger size before ABX and AB 160 injections.
[0071] FIG. 4G shows the ϋ„ and the AUC calculated from the data in FIG. 4F.
[0072] FIG. 4H shows paclitaxel concentrations in the tumors from the second mouse experiment as determined by LC-MS. Data are displayed as μg of paclitaxel/mg of tumor tissue. Student's t-test was utilized to determine if differences were significant.
[0073] FIG. 41 shows 1-125 radioactivity levels in mice treated with AB160 relative to ABX alone.
[0074] FIG. 4J shows a graphical representation of the 1-125 radioactivity levels shown in FIG. 41.
[0075] FIG. 5A shows particle size measurements and affinity of nanoparticles made with rituximab. lO mg/ml of ABX was incubated with rituximab (RIT) at 0-10 mg/ml and light scatter technology (Mastersizer 2000) was used to determine resulting particle sizes. Data are displayed as the percent volume of particles at each size and the curves represent particle size distributions (top). The table (bottom) shows the sizes of the resulting particles at each concentration of antibody.
[0076] FIG. 5B shows particle size measurements and affinity of nanoparticles made with trastuzumab. 10 mg/ml of ABX was incubated with trastuzumab (HER) at 0-22 mg/ml and light scatter technology (Mastersizer 2000) was used to determine resulting particle sizes. Data are displayed as the percent volume of particles at each size and the curves
represent particle size distributions (top). The table (bottom) shows the sizes of the resulting particles at each concentration of antibody.
[0077] FIG. 5C shows the binding affinity of rituximab and trastuzumab as compared to ABX at pH 3, 5, 7 and 9, determined by biolayer interferometry (BLitz) technology. The dissociation constants are displayed for each interaction.
[0078] FIG. 6A shows in vitro toxicity of AR160 as tested with the CD20-positive Daudi human lymphoma cell line. The data are displayed in a graph of the proliferation index, which is the percent of FITC positive cells in treated wells relative to FITC positive cells in the untreated well (the highest level of proliferation).
[0079] FIG. 6B shows in vivo tumor efficacy in athymic nude mice injected with 5 x
6
10 Daudi human lymphoma cells in the right flank. The tumors were allowed to grow
3 3
to 600 mm to 900 mm and the mice were treated with PBS, 30 mg/kg ABX, 12 mg/kg rituximab, 12 mg/kg rituximab + 30 mg/kg ABX, or AR160. Tumor response was determined at day 7 post-treatment by the percent change in tumor size from the first day of treatment. Significance was determined by Student's t-test; the percent change from baseline was significantly different between the AR160 treated mice and all other groups (pO.0001).
[0080] FIG. 6C shows Kaplan-Meier survival curves generated from the experiment shown in FIG. 6B. Median survival for each treatment group is shown. A Mantle-Cox test was used to determine whether survival curve differences were significant.
[0081] FIG. 7A demonstrates addition of another chemotherapy drug (cisplatin) to AB160. ABX (5 mg/ml) was incubated with cisplatin (0.5 mg/ml) at room temperature for 30 minutes and free cisplatin was measured by HPLC in the supernatant after ABX particulate was removed. The quantity of free cisplatin was subtracted from the starting concentration to determine the quantity of cisplatin that bound to the ABX. The data are displayed in a column graph, along with the starting concentration (cisplatin).
[0082] FIG. 7B shows the toxicity of cisplatin-bound ABX (AC) in a proliferation assay of A375 human melanoma cells. After 24 hours of drug exposure and EdU
incorporation, the cells were fixed, permeabilized and labeled with a FITC conjugated anti-EdU antibody. The data is displayed in a graph of the proliferation index, which is
the percent of FITC positive cells in treated wells compared to FITC positive cells in the untreated well (the highest level of proliferation).
[0083] FIG. 7C shows in vivo tumor efficacy of AC (ABC complex; cisplatin-bound
6
ABX) in athymic nude mice injected with 1 x 10 A375 human melanoma cells in the
3 3
right flank. The tumors were allowed to grow to 600 mm to 900 mm and the mice were treated with PBS, 30 mg/kg ABX, 2 mg/kg cisplatin, AB 160, 2 mg/kg cisplatin + AB160 or ABC 160. Tumor response was determined at day 7 post-treatment by the percent change in tumor size from the day of treatment. Significance was determined by Student's t-test; the percent change from baseline was significantly different between the ABC 160 treated mice and PBS-, cisplatin-, or ABX-treated mice (pO.0001). There was no significant difference between the AB160, AB 160 + cisplatin, and ABC160 treated groups for day 7 post-treatment percent change from baseline.
[0084] FIG. 7D shows Kaplan-Meier survival curves generated based on the experiment shown in FIG. 7C and median survival for each treatment group is shown. A Mantle- Cox test was used to determine whether survival curve differences were significant.
[0085] FIG. 8A shows the size distribution of AB160 nanoparticles that were lyophilized, stored at room temperature for one month, and reconstituted, as compared to fresh AB160 or ABX alone.
[0086] FIG. 8B shows the ligand (VEGF) binding ability of AB 160 nanoparticles that were lyophilized, stored at room temperature for one month, and reconstituted, as compared to fresh AB160 or ABX alone.
[0087] FIG. 8C shows in vitro cancer cell toxicity of AB160 nanoparticles that were lyophilized, stored at room temperature for one month, and reconstituted, as compared to fresh AB160 or ABX alone.
[0088] FIG. 8 D shows the size distribution of AB160 nanoparticles that were
lyophilized, stored at room temperature for ten months, and reconstituted, as compared to fresh AB160 or ABX alone.
[0089] FIG. 8 E shows the ligand (VEGF) binding ability of AB160 nanoparticles that were lyophilized, stored at room temperature for ten months, and reconstituted, as compared to fresh AB160 or ABX alone.
[0090] FIG. 8 F shows in vitro cancer cell toxicity of AB160 nanoparticles that were lyophilized, stored at room temperature for ten months, and reconstituted, as compared to fresh AB160 or ABX alone.
[0091] FIGs. 9A-C show the size distributions of the ABX-BEV complexes at IV. infusion conditions (ABX final concentration of 5 mg/mL) incubated in saline at room temperature for up to 24 hours (FIGs. 9A and 9B). By 4 hours at room temperature, there is some evidence of complex breakdown by ELISA (20%, FIG. 9 C).
[0092] FIG. 10 shows in vitro incubation for 30 seconds of ABX (top panel) or AB160 (bottom panel) in saline or heparinized human plasma at relative volume ratios of 9: 1 or 1 : 1.
6
[0093] FIGs. 11 A-E show in vivo testing of athymic nude mice injected with 1 x 10 A375 human melanoma cells in the right flank and treated with (FIG 11 A) PBS, (FIG 11B) 12 mg/kg BEV, (FIG 1 1 C) 30 mg/kg ABX, (FIG 1 1 D) AB 160, or (FIG 1 I E) pretreated with 01.2 mg/kg BEV and, 24hr later, AB160. Data is
3
represented at day 7-post and 10-day treatment as tumor volume in mm .
[0094] FIG. 1 IF summarizes the day 7-post treatment data from FIGs. 1 1 A-E.
[0095] FIG. 11G summarizes the day 1 0-post treatment data from FIGs. 1 1 A-E.
[0096] FIG. 12 depicts the results of an experiment in which CD20 positive Daudi lymphoma cells were labeled with fluorescent tagged anti-human CD20 or isotype matched control in panels F and A, respectively, and analyzed by flow cytometry. In the other panels, the Daudi cells were pretreated with ABRAXANE® (ABX), AR160, AR160L, or Rituxan prior to CD20 labeling. As you can see, CD20 binding was specifically blocked by the AR particles and Rituxan, but not ABX alone suggesting AR160 and AR160L binds their CD20 ligand on these cells blocking binding of the fluorescent anti-CD20.
[0097] FIG. 13 is a histogram overlay of the scatterplots of FIG. 12.
[0098] FIG. 14 depicts a particle size comparison of ABX alone relative to AR and AT freshly made and lyophilized.
[0099] FIG. 15 compares the toxicity of ABX and AR particles in a Daudi cell proliferation assay.
[0100] FIG. 16 depicts the results obtained in mice treated with either labeled
ABRAXANE®, labeled ABRAXANE® coated with non-specific antibodies (AB IgG), or labeled ABRAXANE® coated with Rituximab (AR160). FIG. 16A depicts the fluorescence accumulation in regions of interest (ROI) in tumor (ROI 2, 3, and 4) and in background areas (ROI 1, 5, and 6). ROI 1, 5 and 6 serve as background references. FIG 16B is a bar graph of the average fluorescence per unit of tumor area of mice in all three treatment groups were determined to provide the gross tumor delivery. FIG. 16 C is a bar graph of the average fluorescence per unit of tumor area normalized by background ROI to give proportion of drug delivered to tumor versus body. The data demonstrate that administration of AR160 nanoparticles results in an increased fluorescence as compared to ABRAXANE® alone or ABRAXANE® coated with non-specific antibodies.
[0101] FIG. 17 depicts the survival of the mice treated with a single dose of saline, BEV24 (24 mg/kg), ABX30( 30 mg/kg), AB 160 (12mg/kg BEV and 30mg/kg ABX) and AB225 (24 mg/kg BEV and 30 mg/kg ABS). At 30 days post-administration the survival of mice treated with AB225 and with AB160 far exceeds the survival of mice treated with BEV alone of ABRAXANE® alone.
DETAILED DESCRIPTION
[0102] After reading this description it will become apparent to one skilled in the art how to implement the invention in various alternative embodiments and alternative
applications. However, all the various embodiments of the present invention will not be described herein. It will be understood that the embodiments presented here are presented by way of an example only, and not limitation. As such, this detailed description of various alternative embodiments should not be construed to limit the scope or breadth of the present invention as set forth below.
[0103] Before the present invention is disclosed and described, it is to be understood that the aspects described below are not limited to specific compositions, methods of preparing such compositions, or uses thereof as such may, of course, vary. It is also to be understood that the terminology used herein is for the purpose of describing particular aspects only and is not intended to be limiting.
[0104] The detailed description of the invention is divided into various sections only for the reader's convenience and disclosure found in any section may be combined with that in another section. Titles or subtitles may be used in the specification for the convenience of a reader, which are not intended to influence the scope of the present invention.
Definitions
[0105] Unless defined otherwise, all technical and scientific terms used herein have the same meaning as commonly understood by one of ordinary skill in the art to which this invention belongs. In this specification and in the claims that follow, reference will be made to a number of terms that shall be defined to have the following meanings:
[0106] The terminology used herein is for the purpose of describing particular
embodiments only and is not intended to be limiting of the invention. As used herein, the singular forms "a", "an" and "the" are intended to include the plural forms as well, unless the context clearly indicates otherwise.
[0107] "Optional" or "optionally" means that the subsequently described event or circumstance can or cannot occur, and that the description includes instances where the event or circumstance occurs and instances where it does not.
[0108] The term "about" when used before a numerical designation, e.g., temperature, time, amount, concentration, and such other, including a range, indicates approximations which may vary by ( + ) or ( - ) 10%, 5%,1%, or any subrange or subvalue there between. Preferably, the term "about" when used with regard to a dose amount means that the dose may vary by +/- 10%. For example, "about 400 to about 800 binding agents" indicates that an outside surface of a nanoparticles contain an amount of binding agent between 360 and 880 particles.
[0109] "Comprising" or "comprises" is intended to mean that the compositions and methods include the recited elements, but not excluding others. "Consisting essentially
of when used to define compositions and methods, shall mean excluding other elements of any essential significance to the combination for the stated purpose. Thus, a
composition consisting essentially of the elements as defined herein would not exclude other materials or steps that do not materially affect the basic and novel characteristic(s) of the claimed invention. "Consisting of shall mean excluding more than trace elements of other ingredients and substantial method steps. Embodiments defined by each of these transition terms are within the scope of this invention.
[0110] The term "nanoparticle" as used herein refers to particles having at least one dimension which is less than 5 microns. In preferred embodiments, such as for
intravenous administration, the nanoparticle is less than 1 micron. For direct
administration, the nanoparticle is larger. Even larger particles are expressly contemplated by the invention.
[ 0 1 1 1 ] In a population of particles, the sizes of individual particles are distributed about a mean. Particle sizes for the population can therefore be represented by an average, and also by percentiles. D50 is the particle size below which 50% of the particles fall. 10%) of particles are smaller than the D IO value and 90% of particles are smaller than D90. Where unclear, the "average" size is equivalent to D50. So, for example, AB160 and AR160 refer to nanoparticles having an average size of 160 nanometers.
[ 0 1 1 2 ] The term "nanoparticle" may also encompass discrete multimers of smaller unit nanoparticles. For example, a 320 nm particle comprises a dimer of a unit 160 nm nanoparticle. For 160 nm nanoparticles, multimers would therefore be approximately 320 nm, 480 nm, 640 nm, 800 nm, 960 nm, 1120 nm, and so on.
[0113] The term "carrier protein" as used herein refers to proteins that function to transport binding agents and/or therapeutic agents. The binding agents of the present disclosure can reversibly bind to the carrier proteins. Examples of carrier proteins are discussed in more detail below.
[0114] The term "core" as used herein refers to central or inner portion of the nanoparticle which may be comprised of a carrier protein, a carrier protein and a therapeutic agent, or other agents or combination of agents. In some embodiments, a hydrophobic portion of the binding agent may be incorporated into the core.
[0115] The term "therapeutic agent" as used herein means an agent which is therapeutically useful, e.g., an agent for the treatment, remission or attenuation of a disease state, physiological condition, symptoms, or etiological factors, or for the evaluation or diagnosis thereof. A therapeutic agent may be a chemotherapeutic agent, for example, mitotic inhibitors, topoisomerase inhibitors, steroids, anti-tumor antibiotics,
antimetabolites, alkylating agents, enzymes, proteasome inhibitors, or any combination thereof.
[0116] As used herein, the term, "binding agent", "binding agent specific for", or "binding agent that specifically binds" refers to an agent that binds to a target antigen and does not significantly bind to unrelated compounds. Examples of binding agents that can be effectively employed in the disclosed methods include, but are not limited to, lectins, proteins, and antibodies, such as monoclonal antibodies, e.g. humanized monoclonal antibodies, chimeric antibodies, or polyclonal antibodies, or antigen-binding fragments thereof, as well as aptamers, Fc domain fusion proteins, and aptamers having or fused to hydrophobic protein domain, e.g, Fc domain, etc. In an embodiment the binding agent is an exogenous antibody. An exogenous antibody is an antibody not naturally produced in a mammal, e.g. in a human, by the mammalian immune system.
[0117] The term "antibody" or "antibodies" as used herein refers to immunoglobulin molecules and immunologically active portions of immunoglobulin molecules (i.e., molecules that contain an antigen binding site that immuno-specifically bind an antigen). The term also refers to antibodies comprised of two immunoglobulin heavy chains and two immunoglobulin light chains as well as a variety of forms including full length antibodies and portions thereof; including, for example, an immunoglobulin molecule, a monoclonal antibody, a chimeric antibody, a CDR- grafted antibody, a humanized antibody, a Fab, a Fab', a F(ab')2, a Fv, a disulfide linked Fv, a scFv, a single domain antibody (dAb), a diabody, a multispecific antibody, a dual specific antibody, an anti -idiotypic antibody, a bispecific antibody, a functionally active epitope-binding fragment thereof, bifunctional hybrid antibodies (e.g., Lanzavecchia et al., Eur. J Immunol. 17, 105 (1987)) and single chains (e.g., Huston et al., Proc. Natl. Acad. Sci. USA., 85, 5879-5883 (1988) and Bird et al., Science 242, 423-426 (1988), which are incorporated herein by reference). (See, generally, Hood et al., Immunology, Benjamin, N.Y., 2ND ed. (1984); Harlow and Lane, Antibodies. A Laboratory Manual, Cold Spring Harbor Laboratory (1988); Hunkapiller and
Hood, Nature, 323, 15-16 (1986), which are incorporated herein by reference). The antibody may be of any type (e.g., IgG, IgA, IgM, IgE or IgD). Preferably, the antibody is IgG. An antibody may be non-human (e.g., from mouse, goat, or any other animal), fully human, humanized, or chimeric. Antibody or antibodies include any biosimilar(s) of the antibodies disclosed herein. Biosimilars, as used herein, refers to a biopharmaceutical which is deemed to be comparable in quality, safety, and efficacy to a reference product marketed by an innovator company (Section 351(i) of the Public Health Service Act (42 U.S.C. 262(i)).
[0118] [The term "dissociation constant," also referred to as "]¾," refers to a quantity expressing the extent to which a particular substance separates into individual components (e.g., the protein carrier, antibody, and optional therapeutic agent).
[0119] The terms "lyophilized," "lyophilization" and the like as used herein refer to a process by which the material (e.g., nanoparticles) to be dried is first frozen and then the ice or frozen solvent is removed by sublimation in a vacuum environment. An excipient is optionally included in pre-lyophilized formulations to enhance stability of the lyophilized product upon storage. In some embodiments, the nanoparticles can be formed from lyophilized components (carrier protein, antibody and optional therapeutic) prior to use as a therapeutic. In other embodiments, the carrier protein, binding agent, e.g., antibody, and optional therapeutic agent are first combined into nanoparticles and then lyophilized. The lyophilized sample may further contain additional excipients.
[0120] The term "bulking agents" comprise agents that provide the structure of the fireeze- dried product. Common examples used for bulking agents include mannitol, glycine, lactose and sucrose. In addition to providing a pharmaceutically elegant cake, bulking agents may also impart useful qualities in regard to modifying the collapse temperature, providing freeze-thaw protection, and enhancing the protein stability over long-term storage. These agents can also serve as tonicity modifiers.
[0121] The term "buffer" encompasses those agents which maintain the solution pH in an acceptable range prior to lyophilization and may include succinate (sodium or potassium), histidine, phosphate (sodium or potassium), Tris(tris(hydroxymethyl)aminom ethane), diethanolamine, citrate (sodium) and the like. The buffer of this invention has a pH in the range from about 5.5 to about 6.5; and preferably has a pH of about 6.0. Examples of
buffers that will control the pH in this range include succinate (such as sodium succinate), gluconate, histidine, citrate and other organic acid buffers.
[0122] The term "cryoprotectants" generally includes agents which provide stability to the protein against freezing-induced stresses, presumably by being preferentially excluded from the protein surface. They may al s o offer protection during primary and secondary drying, and long- term product storage. Examples are polymers such as dextran and polyethylene glycol; sugars such as sucrose, glucose, trehalose, and lactose; surfactants such as polysorbates; and amino acids such as glycine, arginine, and serine.
[0123] The term "lyoprotectanf includes agents that provide stability to the protein during the drying or 'dehydration' process (primary and secondary drying cycles), presumably by providing an amorphous glassy m atri x and by binding with the protein through hydrogen bonding, replacing the water molecules that are removed during the drying process. This helps to maintain the protein conformation, minimize protein degradation during the lyophilization cycle and improve the long-term products. Examples include polyols or sugars such as sucrose and trehalose.
[0124] The term "pharmaceutical formulation" refers to preparations which are in such form as to permit the active ingredients t o be effective, and which contains no additional components that are toxic to the subjects to which the formulation would be administered.
[0125] "Pharmaceutically acceptable" excipients (vehicles, additives) are those which can reasonably be administered to a subject mammal to provide an effective dose of the active ingredient employed.
[0126] "Reconstitution time" is the time that is required to rehydrate a lyophilized formulation into a solution.
[0127] A "stable" formulation is one in which the protein therein essentially retains its physical stability and/or chemical stability and/or biological activity upon storage.
[0128] The term "epitope" as used herein refers to the portion of an antigen which is recognized by a binding agent, e.g., an antibody. Epitopes include, but are not limited to, a short amino acid sequence or peptide (optionally glycosylated or otherwise modified) enabling a s p e c i fi c interaction with a protein (e.g., an antibody) or ligand. For example,
an epitope may be a part of a molecule to which the antigen-binding site of a binding agent attaches.
[0129] The term "treating" or "tre atm ent" covers the treatment of a disease or disorder (e.g., cancer), in a subject, such as a human, and includes: (i) inhibiting a disease or disorder, i.e., arresting its development; (ii) relieving a disease or disorder, i.e., causing regression of the disorder; (iii) slowing progression of the disorder; and/or (iv) inhibiting, relieving, or slowing progression of one or more symptoms of the disease or disorder. In some embodiments "treating" or "treatment" refers to the killing of cancer cells.
[0130] The term "kill" with respect to a cancer treatment is directed to include any type of manipulation that will lead to the death of that cancer cell or at least of portion of a population of cancer cells.
[0131] The term "aptamer" refers to a nucleic acid molecule that is capable of binding to a target molecule, such as a polypeptide. For example, an aptamer of the invention can specifically bind to e.g., CD20, CD38, CD52, PD-L1, Ly6E, HER2, HER3/EGFR DAF, ERBB-3 receptor, CSF-1R, STEAP1, CD3, CEA, CD40, OX40, Ang2-VEGF, and VEGF. The generation of antibodies with a particular binding specificity and the therapeutic use of aptamers are well e st ab l i s h e d in the art. See, e.g., U.S. Pat. No. 5,475,096, U.S. Pat. Nos. 5,270,163, 5,582,981, 5,840,867, 6,011,020, 6,051,698, 6, 147,204, 6, 180,348 and 6,699,843, and the therapeutic efficacy of Macugen® (Eyetech, New York) for treating age-related macular degeneration.
[0132] Fc-fusion proteins are bioengineered polypeptides that join the crystallizable fragment (Fc) domain of an antibody with another biologically active agent, e.g., a protein domain, peptide, o r nucleic acid or peptide aptamer to generate a molecule with desired structure- function properties and significant therapeutic potential. The gamma immunoglobulin (IgG) isotype is often used as the basis for generating Fc-fusion proteins because of favorable characteristics such as recruitment of effector function and increased plasma half-life. Given the range of aptamers, both peptide and nucleic acids, that can be used as fusion partners, Fc- fusion proteins have numerous biological and pharmaceutical applications.
[0133] Additionally, some terms used in this specification are more specifically defined below.
Overview
[0134] The current invention is predicated, in part, on the surprising discovery that optionally lyophilized nanoparticles comprising a carrier protein, a binding agent, e.g., an antibody, an aptamer, or a fusion protein having a hydrophobic domain and a binding domain, e.g., an Fc domain fused to an aptamer or the ligand of a cellular receptor, and a therapeutic agent provide targeted therapy to a tumor while minimizing toxicity to the patient. The nanoparticles as described herein are thus a significant improvement versus conventional ADCs.
[0135] For conventional ADCs to be effective, it is critical that the linker be stable enough not to dissociate in the systemic circulation but allow for sufficient drug release at the tumor site. Alley, S.C., et al. (2008) Bioconjug Chem 19:759-765. This has proven to be a major hurdle in developing effective drug conjugate (Julien, D.C., et al. {20\ \) MAbs 3 :467-478; Alley, S.C., et al. (2008) Bioconjug Chem 19:759-765); therefore, an attractive feature of the nano-immune conjugate is that a biochemical linker is not required.
[0136] Another shortcoming of current ADCs is that higher drug penetration into the tumor has not been substantively proven in human tumors. Early testing of ADCs in mouse models suggested that tumor targeting with antibodies would result in a higher concentration of the active agent in the tumor (Deguchi, T. et al. (1986) Cancer Res 46: 3751-3755); however, this has not correlated in the treatment of human disease, likely because human tumors are much more heterogeneous in permeability than mouse tumors. Jain, R.K. et al. (2010) Nat Rev Clin Oncol 7:653-664. Also, the size of the nanoparticle is critical for extravasation from the vasculature into the tumor. In a mouse study using a human colon adenocarcinoma xenotransplant model, the vascular pores were permeable to liposomes up to 400 nm. Yuan, F., et al. (1995) Cancer Res 55: 3752-3756. Another study of tumor pore size and permeability demonstrated that both characteristics were dependent on tumor location and growth status, with regressing tumors and cranial tumors permeable to particles less than 200 nm. Hobbs, S.K., et al. (1998) Proc Natl Acad Sci U SA 95:4607-4612. The nano-immune conjugate described herein overcomes this issue by the fact that the large complex, which is less than 200 nm intact, is partially dissociated in systemic circulation into smaller functional units that are easily able to permeate tumor tissue. Furthermore, once the conjugate arrives to the tumor site, the smaller toxic payload
can be released and only the toxic portion needs to be taken up by tumor cells, not the entire conjugate.
[0137] The advent of antibody- (i.e. AVASTIN®) coated albumin nanoparticles containing a therapeutic agent (i.e., ABRAXA E®) has led to a new paradigm of directional delivery of two or more therapeutic agents to a predetermined site in vivo. See PCT Patent Publication Nos. WO 2012/154861 and WO 2014/055415, each of which is incorporated herein by reference in its entirety.
[0138] When compositions of albumin and an binding agent, e.g., antibody, are admixed together in an aqueous solution at specific concentrations and ratios, the binding agents useful in this invention spontaneously self-assemble into and onto the albumin to form nanoparticles having multiple copies of the binding agent (up to 500 or more). Without being limited to any theory, it is contemplated that the antigen (or ligand) receptor portion of the binding agent, e.g., the antibody or aptamer or Fc fusion molecule, is positioned outward from the nanoparticle while the hydrophobic tail of the binding agent in integrated into the albumin by hydrophobic - hydrophobic interactions.
[0139] While protein compositions comprising a single source protein are commonly stored in lyophilized form where they exhibit significant shelf-life, such lyophilized compositions do not contain a self-assembled nanoparticle of two different proteins integrated together by hydrophobic-hydrophobic interactions. Moreover, the nanoparticle configuration wherein a majority of the binding portions of the binding agent are exposed on the surface of the nanoparticles lends itself to being susceptible to dislodgement or reconfiguration by conditions which otherwise would be considered benign. For example, during
lyophilization, ionic charges on the proteins are dehydrated thereby exposing the underlying charges. Exposed charges allow for charge- charge interactions between the two proteins which can alter the binding affinity of each protein to the other. In addition, the concentration of the nanoparticles increases significantly as the solvent (e.g., water) is removed. Such increased concentrations of nanoparticles could lead to irreversible oligomerization. Oligomerization is a known property of proteins that reduces the biological properties of the oligomer as compared to the monomelic form and increases the size of the particle sometimes beyond 1 micron.
[0140] On the other hand, a stable form of a nanoparticle composition is required for clinical and/or commercial use where a shelf-life of at least 3 months is required and shelf- lives of greater than 6 months or 9 months are preferred. Such a stable composition must be readily available for intravenous injection, must retain its self-assembled form upon intravenous injection so as to direct the nanoparticle to the predetermined site in vivo, must have a maximum size of less than 1 micron so as to avoid any ischemic event when delivered into the blood stream, and finally must be compatible with the aqueous composition used for injection.
Compounds
[0141] As will be apparent to the skilled artisan upon reading this disclosure, the present disclosure relates to compositions of nanoparticles containing a carrier protein, binding agents, and optionally at least one therapeutic agent, wherein said compositions are optionally lyophilized.
[0142] In some embodiments, the carrier protein can be albumin, gelatin, elastin (including topoelastin) or elastin-derived polypeptides (e.g., a-elastin and elastin-like polypeptides (ELPs)), gliadin, legumin, zein, soy protein (e.g., soy protein isolate (SPI)), milk protein (e.g., β-lactoglobulin (BLG) and casein), or whey protein (e.g., whey protein concentrates (WPC) and whey protein isolates (WPI)). In preferred embodiments, the carrier protein is albumin. In preferred embodiments, the albumin is egg white (ovalbumin), bovine serum albumin (BSA), or the like. In even more preferred embodiments, the carrier protein is human serum albumin (HSA). In some embodiments, the carrier protein is a generally regarded as safe (GRAS) excipient approved by the United States Food and Drug
Administration (FDA).
[0143] In some embodiments, the binding agents are antibodies selected from the group consisting of ado- trastuzumab emtansine, alemtuzumab, bevacizumab, cetuximab, denosumab, dinutuximab, ipilimumab, nivolumab, obinutuzumab, ofatumumab, panitumumab, pembrolizumab, pertuzumab, rituximab, and trastuzumab. In some embodiments, the antibodies are a substantially single layer of antibodies on all or part of the surface of the nanoparticle.
Table 1 depicts a list of non-limiting list of antibodies.
Table 1: Antibodies
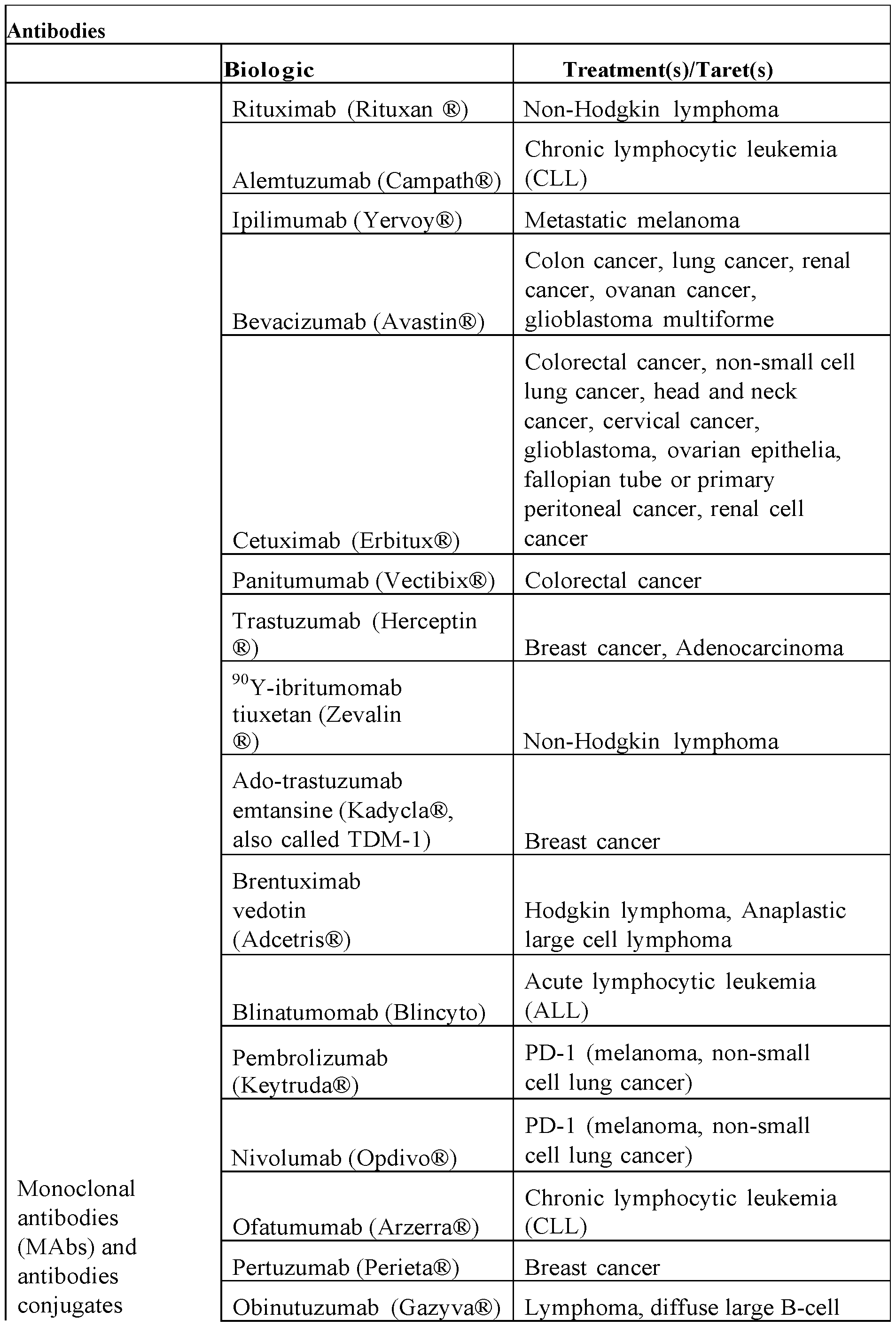

RG7686 (glypican-3 mAb) Hepatocellular carcinoma
HER3 -positive breast cancer, gastric
Perjeta® pertuzumab cancer
[0144] In some embodiments, the at least one therapeutic agent is selected from the group consisting of abiraterone, bendamustine, bortezomib, carboplatin, cabazitaxel, cisplatin, chlorambucil, dasatinib, docetaxel, doxorubicin, epirubicin, erlotinib, etoposide, everolimus, gefitinib, idarubicin, imatinib, hydroxyurea, imatinib, lapatinib, leuprorelin, melphalan, methotrexate, mitoxantrone, nedaplatin, nilotinib, oxaliplatin, paclitaxel, pazopanib, pemetrexed, picoplatin, romidepsin, satraplatin, sorafenib, vemurafenib, sunitinib, teniposide, triplatin, vinblastine, vinorelbine, vincristine, and cyclophosphamide.
[0145] Table 2 depicts a list of non-limiting list of cancer therapeutic agents.
Table 2: Cancer therapeutic agents

Adrucil (Fluorouracil) Basal cell carcinoma; breast cancer;
colorectal cancer; gastric (stomach) adenocarcinoma; pancreatic cancer; squamous cell carcinoma of the head and neck
Afatinib Dimaleate Non-small cell lung cancer
Afinitor (Everolimus) Breast cancer, pancreatic cancer; renal
cell carcinoma; subependymal giant cell astrocytoma
Alimta (Pemetrexed Disodium) Malignant pleural mesothelioma; non- small cell lung cancer
Ambochlorin (Chlorambucil) Chronic lymphocytic leukemia;
Hodgkin lymphoma; non-Hodgkin
Anastrozole Breast cancer
Aredia (Pamidronate Disodium) Breast cancer; multiple myeloma
Arimidex (Anastrozole) Breast cancer
Aromasin (Exemestane) Advanced breast cancer; early-stage
breast cancer and estrogen receptor
Arranon (Nelarabine) T-cell acute lymphoblastic leukemia; T- cell lymphoblastic lymphoma
Azacitidine Myelodysplastic syndromes
BEACOPP Hodgkin lymphoma
Becenum (Carmustine) Brain tumors; Hodgkin lymphoma;
multiple myeloma; non-Hodgkin
Beleodaq (Belinostat) Peripheral T-cell lymphoma
BEP Ovarian germ cell tumors; testicular germ cell tumors
Bicalutamide Prostate cancer
BiCNU (Carmustine) Brain tumors; Hodgkin lymphoma;
multiple myeloma; non-Hodgkin
Bleomycin Hodgkin lymphoma; non-Hodgkin
lymphoma; penile cancer; squamous cell carcinoma of the cervix; squamous cell carcinoma of the head and neck; squamous cell carcinoma of the vulva; testicular cancer
Bosulif (Bosutinib) Chronic myelogenous leukemia
Brentuximab Vedotin Anaplastic large cell lymphoma;
Hodgkin lymphoma
Busulfan Chronic myelogenous leukemia
Busulfex (Busulfan) Chronic myelogenous leukemia
Cabozantinib-S-Malate Medullary thyroid cancer
CAF Breast cancer
Camptosar (Irinotecan Hydrochloride) Colorectal cancer
CAPOX Colorectal cancer
Carfilzomib Multiple myeloma
Casodex (Bicalutamide) Prostate cancer
CeeNU (Lomustine) Brain tumors; Hodgkin lymphoma
Ceritinib Non-small cell lung cancer
Cerubidine (Daunorubicin Hydrochloride) Acute lymphoblastic leukemia; acute myeloid leukemia
Chlorambucil Chronic lymphocytic leukemia;
Hodgkin lymphoma; non-Hodgkin
CHLORAMBUCIL-PREDNISONE Chronic lymphocytic leukemia
CHOP Non-Hodgkin lymphoma
Cisplatin Bladder cancer; cervical cancer; malignant mesothelioma; non-small cell lung cancer; ovarian cancer; squamous cell carcinoma of the head and neck; testicular cancer
Clafen (Cyclophosphamide) Acute lymphoblastic leukemia; acute
myeloid leukemia; breast cancer; chronic lymphocytic leukemia; chronic myelogenous leukemia; Hodgkin lymphoma; multiple myeloma; mycosis fungoides;
neuroblastoma; non- Hodgkin lymphoma; ovarian cancer; retinoblastoma
Clofarex (Clofarabine) Acute lymphoblastic leukemia
CMF Breast cancer
Cometriq ( Cabozantinib-S-Malate) Medullary thyroid cancer
COPP Hodgkin lymphoma; non-Hodgkin lymphoma
COPP-ABV Hodgkin lymphoma
Cosmegen (Dactinomycin) Ewing sarcoma; gestational trophoblastic disease; rhabdomyosarcoma; solid tumors; testicular cancer; Wilms tumor
CVP Non-Hodgkin lymphoma; chronic
lymphocytic leukemia
Cyclophosphamide Acute lymphoblastic leukemia; acute
myeloid leukemia; breast cancer; chronic lymphocytic leukemia; chronic myelogenous leukemia; Hodgkin lymphoma; multiple myeloma; mycosis fungoides;
neuroblastoma; non- Hodgkin lymphoma; ovarian cancer; retinoblastoma.
Cyfos (Ifosfamide) Testicular germ cell tumors
Cyramza (Ramucirumab) Adenocarcinoma; colorectal cancer; non- small cell lung cancer
Cytarabine Acute lymphoblastic leukemia; acute
myeloid leukemia; chronic myelogenous leukemia; meningeal leukemia
Cytosar-U (Cytarabine) Acute lymphoblastic leukemia; acute
myeloid leukemia; chronic myelogenous leukemia; meningeal leukemia
Cytoxan (Cyclophosphamide) Acute lymphoblastic leukemia; acute myeloid leukemia; breast cancer; chronic lymphocytic leukemia; chronic myelogenous
Hodgkin lymphoma; multiple
myeloma; mycosis fungoides;
neuroblastoma; non- Hodgkin
lymphoma; ovarian cancer;
Dacarbazine Hodgkin lymphoma; melanoma
Dacogen (Decitabine) Myelodysplastic syndromes
Dactinomycin Ewing sarcoma; gestational trophoblastic disease; rhabdomyosarcoma; solid
tumors; testicular cancer; Wilms tumor
Daunorubicin Hydrochloride Acute lymphoblastic leukemia; acute
myeloid leukemia
Degarelix Prostate cancer
Denileukin Diftitox Cutaneous T-cell lymphoma
Denosumab Giant cell tumor of the bone; breast
cancer, prostate cancer
DepoCyt (Liposomal Cytarabine) Lymphomatous meningitis
DepoFoam (Liposomal Cytarabine) Lymphomatous meningitis
Docetaxel Breast cancer; adenocarcinoma of the
stomach or gastroesophageal junction; non- small cell lung cancer; prostate cancer;
squamous cell carcinoma of the head and
Doxil (Doxorubicin Hydrochloride Liposome) AIDS-related Kaposi sarcoma;
multiple myeloma; ovarian cancer
Doxorubicin Hydrochloride Acute lymphoblastic leukemia; acute myeloid leukemia; breast cancer; gastric (stomach) cancer; Hodgkin lymphoma; neuroblastoma; non-Hodgkin lymphoma; ovarian cancer; small cell lung cancer; soft tissue and bone sarcomas; thyroid cancer; transitional cell bladder cancer; Wilms tumor.
Dox-SL (Doxorubicin AIDS-related Kaposi sarcoma;
Hydrochloride Liposome) multiple myeloma; ovarian cancer
DTIC-Dome (Dacarbazine) Hodgkin lymphoma; melanoma
Efudex (Fluorouracil) Basal cell carcinoma; breast cancer;
colorectal cancer; gastric (stomach) adenocarcinoma; pancreatic cancer; squamous cell carcinoma of the head and neck
Ellence (Epirubicin Hydrochloride) Breast cancer
Eloxatin (Oxaliplatin) Colorectal cancer; stage III colon cancer
Emend (Aprepitant) Nausea and vomiting caused by
chemotherapy and nausea and vomiting after
Enzalutamide Prostate cancer
Epirubicin Hydrochloride Breast cancer
EPOCH Non-Hodgkin lymphoma
Erbitux (Cetuximab) Colorectal cancer; squamous cell carcinoma of the head and neck
Eribulin Mesylate Breast cancer
Erivedge (Vismodegib) Basal cell carcinoma
Erlotinib Hydrochloride Non-small cell lung cancer; pancreatic cancer
Erwinaze (Asparaginase Acute lymphoblastic leukemia
Erwinia chrysanthemi)
Etopophos (Etoposide Phosphate) Small cell lung cancer; testicular cancer
Evacet (Doxorubicin Hydrochloride Liposome) AIDS-related Kaposi sarcoma;
multiple myeloma; ovarian cancer
Everolimus Breast cancer; pancreatic cancer; renal
cell carcinoma; subependymal giant cell astrocytoma
Evista (Raloxifene Hydrochloride) Breast cancer
Exemestane Breast cancer
Fareston (Toremifene) Breast cancer
Farydak (Panobinostat) Multiple myeloma
Faslodex (Fulvestrant) Breast cancer
FEC Breast cancer
Femara (Letrozole) Breast cancer
Filgrastim Neutropenia
Fludara (Fludarabine Phosphate) Chronic lymphocytic leukemia
Fluoroplex (Fluorouracil) Basal cell carcinoma; breast cancer;
colorectal cancer; gastric (stomach) adenocarcinoma; pancreatic cancer; squamous cell carcinoma of the head and neck
Folex (Methotrexate) Acute lymphoblastic leukemia; breast
cancer; gestational trophoblastic disease; head and neck cancer; lung cancer; mycosis fungoides; non-Hodgkin lymphoma;
FOLFIRI Colorectal cancer
FOLFIRI-BEVACIZUMAB Colorectal cancer
FOLFIRI-CETUXIMAB Colorectal cancer
FOLFIRINOX Pancreatic cancer
FOLFOX Colorectal cancer
Folotyn (Pralatrexate) Peripheral T-cell lymphoma
FU-LV Colorectal cancer; esophageal cancer;
gastric cancer
Fulvestrant Breast cancer
Gefitinib Non-small cell lung cancer
Gemcitabine Hydrochloride Breast cancer; non-small cell lung
cancer; ovarian cancer; pancreatic cancer
GEMCITABINE-CISPLATIN Biliary tract cancer; bladder cancer;
cervical cancer; malignant mesothelioma; non-small cell lung cancer; ovarian cancer; pancreatic cancer
GEMCITABINE-OXALIPLATIN Pancreatic cancer
Gemtuzumab Ozogamicin (antibody drug Acute myeloid leukemia
conjugate)
Gemzar (Gemcitabine Hydrochloride) Breast cancer; non-small cell lung
cancer; ovarian cancer; pancreatic cancer
Gilotrif (Afatinib Dimaleate) Non-small cell lung cancer
Gleevec (Imatinib Mesylate) Acute lymphoblastic leukemia; chronic
eosinophilic leukemia or hypereosinophilic syndrome; chronic myelogenous leukemia; dermatofibrosarcoma protuberans;
gastrointestinal stromal tumor;
myelodysplastic/myeloproliferative neoplasms; systemic mastocytosis.
Gliadel (Carmustine Implant) Glioblastoma multiforme; malignant glioma
Goserelin Acetate Breast cancer; prostate cancer
Halaven (Eribulin Mesylate) Breast cancer
Hycamtin (Topotecan Hydrochloride) Cervical cancer; ovarian cancer; small cell lung cancer
Hyper-CVAD Acute lymphoblastic leukemia; non- Hodgkin lymphoma
Ibrance (Palbociclib) Breast cancer
Ibrutinib Chronic lymphocytic leukemia; mantel
cell lymphoma;
ICE Hodgkin lymphoma; non-Hodgkin lymphoma
Iclusig (Ponatinib Hydrochloride) Acute lymphoblastic leukemia; Chronic
myelogenous leukemia
Idamycin (Idarubicin Hydrochloride) Acute myeloid leukemia
Imatinib Mesylate Acute lymphoblastic leukemia; chronic
eosinophilic leukemia or hypereosinophilic syndrome; chronic myelogenous leukemia; dermatofibrosarcoma protuberans;
gastrointestinal stromal tumor;
myelodysplastic/myeloproliferative neoplasms; systemic mastocytosis.
Imbruvica (Ibrutinib) Chronic lymphocytic leukemia; mantle cell lymphoma; Waldenstr6m
Inlyta (Axitinib) Renal cell carcinoma
Iressa (Gefitinib) Non-small cell lung cancer
Irinotecan Hydrochloride Colorectal cancer
Istodax (Romidepsin) Cutaneous T-cell lymphoma
Ixempra (Ixabepilone) Breast cancer
Jevtana (Cabazitaxel) Prostate cancer
Keoxifene (Raloxifene Hydrochloride) Breast cancer
Kyprolis (Carfilzomib) Multiple myeloma
Lenvima (Lenvatinib Mesylate) Thyroid cancer
Letrozole Breast cancer
Leucovorin Calcium Colorectal cancer
Leukeran (Chlorambucil) Chronic lymphocytic leukemia;
Hodgkin lymphoma; non-Hodgkin
Leuprolide Acetate Prostate cancer
Linfolizin (Chlorambucil) Chronic lymphocytic leukemia;
Hodgkin lymphoma; non-Hodgkin
LipoDox (Doxorubicin AIDS-related Kaposi sarcoma;
Hydrochloride Liposome) multiple myeloma; ovarian cancer
Lomustine Brain tumors; Hodgkin lymphoma
Lupron (Leuprolide Acetate) Prostate cancer
Lynparza (Olaparib) Ovarian cancer
Marqibo (Vincristine Sulfate Liposome) Acute lymphoblastic leukemia
Matulane (Procarbazine Hydrochloride) Hodgkin lymphoma
Mechlorethamine Hydrochloride Bronchogenic carcinoma; chronic
lymphocytic leukemia; chronic myelogenous leukemia; Hodgkin lymphoma; malignant pleural effusion, malignant pericardial effusion, and malignant peritoneal effusion; mycosis fungoides; non-Hodgkin lymphoma
Megace (Megestrol Acetate) Breast cancer; endometrial cancer
Mekinist (Trametinib) Melanoma
Mercaptopurine Acute lymphoblastic leukemia
Mesnex (Mesna) Hemorrhagic cystitis
Methazolastone (Temozolomide) Anaplastic astrocytoma;
glioblastoma multiforme
Mexate (Methotrexate) Acute lymphoblastic leukemia; breast
cancer; gestational trophoblastic disease; head and neck cancer; lung cancer; mycosis fungoides; non-Hodgkin lymphoma;
Mexate-AQ (Methotrexate) Acute lymphoblastic leukemia; breast
cancer; gestational trophoblastic disease; head and neck cancer; lung cancer; mycosis fungoides; non-Hodgkin lymphoma;
Mitoxantrone Hydrochloride Acute myeloid leukemia; prostate cancer
Mitozytrex (Mitomycin C) Gastric (stomach) and
pancreatic adenocarcinoma
MOPP Hodgkin lymphoma
Mozobil (Plerixafor) Multiple myeloma; non-Hodgkin lymphoma
Mustargen (Mechlorethamine Hydrochloride) Bronchogenic carcinoma; chronic
lymphocytic leukemia; chronic myelogenous leukemia; Hodgkin lymphoma; malignant pleural effusion, malignant pericardial effusion, and malignant peritoneal effusion; mycosis fungoides; non-Hodgkin lymphoma
Myleran (Busulfan) Chronic myelogenous leukemia
Mylotarg (Gemtuzumab Ozogamicin) Acute myeloid leukemia
Nanoparticle Paclitaxel (Paclitaxel Albumin- Breast cancer; Non-small cell lung stabilized Nanoparticle Formulation) cancer; Pancreatic cancer
Navelbine (Vinorelbine Tartrate) Non-small cell lung cancer
Nelarabine T-cell acute lymphoblastic leukemia
Neosar (Cyclophosphamide) Acute lymphoblastic leukemia; Acute
myeloid leukemia; Breast cancer; Chronic lymphocytic leukemia; Chronic myelogenous leukemia; Hodgkin lymphoma; Multiple myeloma; Mycosis fungoides;
Neuroblastoma; Non- Hodgkin lymphoma; Ovarian cancer; Retinoblastoma
Nexavar (Sorafenib Tosylate) Hepatocellular carcinoma; Renal
cell carcinoma; Thyroid cancer
Nilotinib Chronic myelogenous leukemia
Nivolumab Melanoma; Squamous non-small cell
lung cancer
Nolvadex (Tamoxifen Citrate) Breast cancer
Odomzo (Sonidegib) Basal cell carcinoma
OEPA Hodgkin lymphoma
OFF Pancreatic cancer
Olaparib Ovarian cancer
Oncaspar (Pegaspargase) Acute lymphoblastic leukemia
OPPA Hodgkin lymphoma
Oxaliplatin Colorectal cancer; Stage III colon cancer
Paclitaxel AIDS-related Kaposi sarcoma; Breast
cancer; Non-small cell lung cancer; Ovarian
Paclitaxel Albumin-stabilized Nanoparticle Breast cancer; Non-small lung
Formulation cancer; Pancreatic cancer
PAD Multiple myeloma
Palbociclib Breast cancer
Pamidronate Disodium Breast cancer; Multiple myeloma
Panitumumab Colorectal cancer
Panobinostat Multiple myeloma
Paraplat (Carboplatin) Non-small cell lung cancer; Ovarian cancer
Paraplatin (Carboplatin) Non-small cell lung cancer; Ovarian cancer
Pazopanib Hydrochloride Renal cell carcinoma; Soft tissue sarcoma
Pegaspargase Acute lymphoblastic leukemia
Pemetrexed Disodium Malignant pleural mesothelioma; Non- small cell lung cancer
Platinol (Cisplatin) Bladder cancer; Cervical cancer; Malignant mesothelioma; Non-small cell lung cancer; Ovarian cancer; Squamous cell carcinoma of the head and neck; Testicular cancer
Platinal-AQ (Cisplatin) Bladder cancer; Cervical cancer; Malignant mesothelioma; Non-small cell lung cancer; Ovarian cancer; Squamous cell carcinoma of the head and neck; Testicular cancer
Plerixafor Multiple myeloma; Non-Hodgkin lymphoma
Pomalidomide Multiple myeloma
Pomalyst (Pomalidomide) Multiple myeloma
Pontinib Hydrochloride Acute lymphoblastic leukemia;
Chronic myelogenous leukemia
Pralatrexate Peripheral T-cell lymphoma
Prednisone Acute lymphoblastic leukemia; Chronic lymphocytic leukemia; Hodgkin lymphoma; Multiple myeloma; Non-Hodgkin lymphoma; Prostate cancer; Thymoma and thymic carcmoma
Procarbazine Hydrochloride Hodgkin lymphoma
Provenge (Sipuleucel-T) Prostate cancer
Purinethol (Mercaptopurine) Acute lymphoblastic leukemia
Radium 223 Dichloride Prostate cancer
Raloxifene Hydrochloride Breast cancer
R-CHOP Non-Hodgkin lymphoma
R-CVP Non-Hodgkin lymphoma
Regorafenib Colorectal cancer; Gastrointestinal
stromal tumor
R-EPOCH B-cell non-Hodgkin lymphoma
Revlimid (Lenalidomide) Mantle cell lymphoma; Multiple
myeloma; Anemia
Rheumatrex (Methotrexate) Acute lymphoblastic leukemia; Breast
cancer; Gestational trophoblastic disease; Head and neck cancer; Lung cancer; Non- Hodgkin lymphoma; Osteosarcoma
Romidepsin Cutaneous T-cell lymphoma
Rubidomycin (Daunorubicin Hydrochloride) Acute lymphoblastic leukemia; Acute
myeloid leukemia
Sipuleucel-T Prostate cancer
Somatuline Depot (Lanreotide Acetate) Gastroenteropancreatic neuroendocrine tumors
Sonidegib Basal cell carcinoma
Sorafenib Tosylate Hepatocellular carcinoma; Renal
cell carcinoma; Thyroid cancer
Sprycel (Dasatinib) Acute lymphoblastic leukemia;
Chronic myelogenous leukemia
STANFORD V Hodgkin lymphoma
Stivarga (Regorafenib) Colorectal cancer; Gastrointestinal
stromal tumor
Sunitnib Malate Gastronintestinal stromal tumor; Pancreatic cancer; Renal cell carcinoma
Sutent (Sunitinib Malate) Gastronintestinal stromal tumor;
Pancreatic cancer; Renal cell carcinoma
Synovir (Thalidomide) Multiple myeloma
Synribo (Omacetaxine Mepesuccinate) Chronic myelogenous leukemia
TAC Breast cancer
Tafinlar (Dabrafenib) Melanoma
Tamoxifen Citrate Breast cancer
Tarabine PFS (Cytarabine) Acute lymphoblastic leukemia; Acute
myeloid leukemia; Chronic myelogenous
Tarceva (Erlotinib Hydrochloride) Non-small cell lung cancer; Pancreatic cancer
Targretin (Bexarotene) Skin problems caused by cutaneous T- cell lymphoma
Tasigna (Niltinib) Chronic myelogenous leukemia
Taxol (Paclitaxel) AIDS-related Kaposi sarcoma; Breast
cancer; Non-small cell lung cancer; Ovarian
Taxotere (Docetaxel) Breast cancer; Adenocarcinoma; Non- small cell lung cancer; Prostate cancer; Squamous cell carcinoma of the head and
Temodar (Temozolomide) Anaplastic astrocytoma;
Glioblastoma multiforme
Temozolomide Anaplastic astrocytoma;
Glioblastoma multiforme
Thiotepa Bladder cancer; Breast cancer; Malignant pleural effusion, malignant pericardial effusion, and malignant peritoneal effusion; Ovarian cancer
Toposar (Etoposide) Small cell lung cancer; Testicular cancer
Topotecan Hydrochloride Cervical cancer; Ovarian cancer; Small cell lung cancer
Toremifene Breast cancer
Torisel (Temsirolimus) Renal cell carcinoma
TPF Squamous cell carcinoma of the head and neck; Gastric (stomach) cancer
Trastuzumab Adenocarcinoma; Breast cancer
Treanda (Bendamustine Hydrochloride) B-cell non-Hodgkin lymphoma;
Chronic lymphocytic leukemia
Trisenox (Arsenic Trioxide) Acute promyelocytic leukemia
Tykerb (Lapatinib Ditosylate) Breast cancer
Vandetabib Medullary thyroid cancer
VAMP Hodgkin lymphoma
VelP Ovarian germ cell; Testicular cancer
Velban (Vinblastine Sulfate) Breast cancer; Choriocarcinoma;
Hodgkin lymphoma; Kaposi sarcoma; Mycosid fungoides; Non-Hodgkin
Testicular cancer
Velcade (Bortezomib) Mulitple myeloma; Mantle cell lymphoma
Velsar (Vinblastine Sulfate) Breast cancer; Choriocarcinoma;
Hodgkin lymphoma; Kaposi sarcoma; Mycosis fungoides; Non-Hodgkin lymphoma; Testicular cancer
VePesid (Etoposide) Small cell lung cancer; Testicular cancer
Viadur (Leuprolide Acetate) Prostate cancer
Vidaza (Azacitidine) Myelodysplastic syndromes
Vincasar PFS (Vincristine Sulfate) Acute leukemia; Hodgkin lymphoma;
Neuroblastoma; Non-Hodgkin
lymphoma; Rhabdomyosarcoma; Wilms
Vincristine Sulfate Liposome Acute lymphoblastic leukemia
Vinorelbine Tartrate Non-small cell lung cancer
VIP Testicular cancer
Visbodegib Basal cell carcinoma
Voraxaze (Glucarpidase) Toxic blood levels of the anticancer drug methotrexate
Votrient (Pazopanib Hydrochloride) Renal cell carcinoma; Soft tissue sarcoma
Wellcovorin (Leucovorin Calcium) Colorectal cancer; Anemia
Xalkori Crizotinib) Non-small cell lung cancer
Xeloda Capecitabine) Breast cancer; Colorectal cancer
XELIRI Colorectal cancer; Esophageal cancer;
Gastric (stomach) cancer
XELOX Colorectal cancer
Xofigo (Radium 223 Dichloride) Prostate cancer
Xtandi (Enzalutamide) Prostate cancer
Zaltrap (Ziv-Aflibercept) Colorectal cancer
Zelboraf (Vemurafenib) Melanoma
Ziv-Aflibercept Colorectal cancer
Zoladex (Goserelin Acetate) Breast cancer; Prostate cancer
Zolinza (Vorinostat) Cutaneous T-cell lymphoma
Zometa (Zoledronic Acid) Multiple myeloma
Zydelig (Idelalisib) Chronic lymphocytic leukemia; Non- Hodgkin lymphoma (Follicula B-cell non Hodgkin lymphoma and Small lymphocytic
Zykadia (Certinib) Non-small cell lung cancer
Zytiga (Abiraterone Acetate) Prostate cancer
[0146] It is to be understood that the therapeutic agent may be located inside the nanoparticle, on the outside surface of the nanoparticle, or both. The nanoparticle may contain more than one therapeutic agent, for example, two therapeutic agents, three therapeutic agents, four therapeutic agents, five therapeutic agents, or more. Furthermore, a nanoparticle may contain the same or different therapeutic agents inside and outside the nanoparticle.
[0147] In some embodiments of this invention the nanoparticles comprising ABRAXA E and bevacizumab are excluded.
[0148] In one aspect, the nanoparticle comprises at least 100 binding agents non-covalently bound to the surface of the nanoparticle. In one aspect, the nanoparticle comprises at least 200 binding agents non-covalently bound to the surface of the nanoparticle. In one aspect, the nanoparticle comprises at least 300 binding agents non-covalently bound to the surface of the nanoparticle. In one aspect, the nanoparticle comprises at least 400 binding agents non-covalently bound to the surface of the nanoparticle. In one aspect, the nanoparticle comprises at least 500 binding agents non-covalently bound to the surface of the nanoparticle. In one aspect, the nanoparticle comprises at least 600 binding agents non- covalently bound to the surface of the nanoparticle.
[0149] In one aspect, the nanoparticle comprises between about 100 and about 1000 binding agents non-covalently bound to the surface of the nanoparticle. In one aspect, the nanoparticle comprises between about 200 and about 1000 binding agents non-covalently bound to the surface of the nanoparticle. In one aspect, the nanoparticle comprises between about 300 and about 1000 binding agents non-covalently bound to the surface of
the nanoparticle. In one aspect, the nanoparticle comprises between about 400 and about 1000 binding agents non-covalently bound to the surface of the nanoparticle. In one aspect, the nanoparticle comprises between about 500 and about 1000 binding agents non- covalently bound to the surface of the nanoparticle. In one aspect, the nanoparticle comprises between about 600 and about 1000 binding agents non-covalently bound to the surface of the nanoparticle. In one aspect, the nanoparticle comprises between about 200 and about 800 binding agents non-covalently bound to the surface of the nanoparticle. In one aspect, the nanoparticle comprises between about 300 and about 800 binding agents non-covalently bound to the surface of the nanoparticle. In preferred embodiments, the nanoparticle comprises between about 400 and about 800 binding agents non-covalently bound to the surface of the nanoparticle. Contemplated values include any value or subrange within any of the recited ranges, including endpoints.
[0150] In one aspect, the average particle size in the nanoparticle composition is less than about 1 μιη. In one aspect, the average particle size in the nanoparticle composition is between about 130 nm and about 1 μιη. In one aspect, the average particle size in the nanoparticle composition is between about 130 nm and about 900 nm. In one aspect, the average particle size in the nanoparticle composition is between about 130 nm and about 800 nm. In one aspect, the average particle size in the nanoparticle composition is between about 130 nm and about 700 nm. In one aspect, the average particle size in the
nanoparticle composition is between about 130 nm and about 600 nm. In one aspect, the average particle size in the nanoparticle composition is between about 130 nm and about 500 nm. In one aspect, the average particle size in the nanoparticle composition is between about 130 nm and about 400 nm. In one aspect, the average particle size in the
nanoparticle composition is between about 130 nm and about 300 nm. In one aspect, the average particle size in the nanoparticle composition is between about 130 nm and about 200 nm. In a preferred embodiment, the average particle size in the nanoparticle composition is between about 150 nm and about 180 nm. In an especially preferred embodiment, the mean particle size in the nanoparticle composition is about 160 nm. Contemplated values include any value, subrange, or range within any of the recited ranges, including endpoints.
[0151] In one aspect, the nanoparticle composition is formulated for intravenous injection. In order to avoid an ischemic event, the nanoparticle composition formulated for
intravenous injection should comprise nanoparticles with an average particle size of less than about 1 μιη.
[0152] In one aspect, the average particle size in the nanoparticle composition is greater than about 1 μιη. In one aspect, the average particle size in the nanoparticle composition is between about 1 μιη and about 5 μιη. In one aspect, the average particle size in the nanoparticle composition is between about 1 μιη and about 4 μιη. In one aspect, the average particle size in the nanoparticle composition is between about 1 μιη and about 3 μιη. In one aspect, the average particle size in the nanoparticle composition is between about 1 μπι and about 2 μιη. In one aspect, the average particle size in the nanoparticle composition is between about 1 μιη and about 1.5 μιη. Contemplated values include any value, subrange, or range within any of the recited ranges, including endpoints.
[0153] In one aspect, the nanoparticle composition is formulated for direct injection into a tumor. Direct injection includes injection into or proximal to a tumor site, perfusion into a tumor, and the like. When formulated for direct injection into a tumor, the nanoparticle may comprise any average particle size. Without being bound by theory, it is believed that larger particles (e.g., greater than 500 nm, greater than 1 μπι, and the like) are more likely to be immobilized within the tumor, thereby providing a beneficial effect. Larger particles can accumulate in the tumor or specific organs. See, e.g., 20-60 micron glass particle that is used to inject into the hepatic artery feeding a tumor of the liver, called "TheraSphere®" (in clinical use for liver cancer). Therefore, for intravenous administration, particles under 1 μπι are typically used. Particles over 1 μπι are, more typically, administered directly into a tumor ("direct injection") or into an artery feeding into the site of the tumor.
[0154] In one aspect, less than about 0.01% of the nanoparticles within the composition have a particle size greater than 200 nm, greater than 300 nm, greater than 400 nm, greater than 500 nm, greater than 600 nm, greater than 700 nm, or greater than 800 nm. In one aspect, less than about 0.001% of the nanoparticles within the composition have a particle size greater than 200 nm, greater than 300 nm, greater than 400 nm, greater than 500 nm, greater than 600 nm, greater than 700 nm, or greater than 800 nm. In a preferred embodiment, less than about 0.01% of the nanoparticles within the composition have a particle size greater than 800 nm. In a more preferred embodiment, less than about 0.001%) of the nanoparticles within the composition have a particle size greater than 800 nm.
[0155] In a preferred aspect, the sizes and size ranges recited herein relate to particle sizes of the reconstituted lyophilized nanoparticle composition. That is, after the lyophilized nanoparticles are resuspended in an aqueous solution (e.g., water, other pharmaceutically acceptable excipient, buffer, etc.), the particle size or average particle size is within the range recited herein.
[0156] In one aspect, at least about 50%, 60%, 70%, 80%, 90%, 95%, 96%, 97%, 98%, 99%), 99.5%), or 99.9% of the nanoparticles are present in the reconstituted composition as single nanoparticles. That is, fewer than about 50%, 40%, 30%, etc. of the nanoparticles are dimerized or multimerized (oligomerized).
[0157] In some embodiments, the size of the nanoparticle can be controlled by the adjusting the amount (e.g., ratio) of carrier protein to binding agent. The size of the nanoparticles, and the size distribution, is also important. The nanoparticles of the invention may behave differently according to their size. At large sizes, an agglomeration may block blood vessels. Therefore, agglomeration of nanoparticles can affect the performance and safety of the composition. On the other hand, larger particles may be more therapeutic under certain conditions (e.g., when not administered intravenously).
[0158] In one aspect, the nanoparticle composition comprises at least one additional therapeutic agent. In one embodiment, the at least one additional therapeutic agent is non- covalently bound to the outside surface of the nanoparticle. In one embodiment, the at least one additional therapeutic agent is arranged on the outside surface of the nanoparticle. In one embodiment, the at least one additional therapeutic agent is selected from the group consisting of abiraterone, bendamustine, bortezomib, carboplatin, cabazitaxel, cisplatin, chlorambucil, dasatinib, docetaxel, doxorubicin, epirubicin, erlotinib, etoposide, everolimus, gemcitabine, gefitinib, idarubicin, imatinib, hydroxyurea, imatinib, lapatinib, leuprorelin, melphalan, methotrexate, mitoxantrone, nedaplatin, nilotinib, oxaliplatin, pazopanib, pemetrexed, picoplatin, romidepsin, satraplatin, sorafenib, vemurafenib, sunitinib, teniposide, triplatin, vinblastine, vinorelbine, vincristine, and cyclophosphamide. In one embodiment, the at least one additional therapeutic agent is an anti- cancer binding agent, e.g., an anti-cancer antibody.
Methods of Making Nanoparticles
[0159] In some aspects, the current invention relates to methods of making nanoparticle compositions as described herein.
[0160] In one aspect, the nanoparticles of the nanoparticle composition are formed by contacting the carrier protein or carrier protein-therapeutic agent particle with the binding agent at a ratio of about 10: 1 to about 10:30 carrier protein particle or carrier protein- therapeutic agent particle to binding agent. In one embodiment, the ratio is about 10:2 to about 10:25. In one embodiment, the ratio is about 10:2 to about 1 : 1. In a preferred embodiment, the ratio is about 10:2 to about 10:6. In an especially preferred embodiment, the ratio is about 10:4. Contemplated ratios include any value, subrange, or range within any of the recited ranges, including endpoints.
[0161] In one embodiment, the amount of solution or other liquid medium employed to form the nanoparticles is particularly important. No nanoparticles are formed in an overly dilute solution of the carrier protein (or carrier protein-therapeutic agent) and the antibodies. An overly concentrated solution will result in unstructured aggregates. In some embodiments, the amount of solution (e.g., sterile water, saline, phosphate buffered saline) employed is between about 0.5 mL of solution to about 20 mL of solution. In some embodiments, the amount of carrier protein is between about 1 mg/mL and about lOO mg/mL. In some embodiments, the amount of binding agent is between about 1 mg/mL and about 30 mg/mL. For example, in some embodiments, the ratio of carrier proteimbinding agent: solution is approximately 9 mg of carrier protein (e.g., albumin) to 4 mg of binding agent, e.g., antibody (e.g., BEV) in 1 mL of solution (e.g., saline). An amount of therapeutic agent (e.g., taxol) can also be added to the carrier protein. For example, 1 mg of taxol can be added 9 mg of carrier protein (10 mg carrier protein- therapeutic) and 4 mg of binding agent, e.g., antibody, Fc fusion molecule, or aptamer, in 1 mL of solution. When using a typical i.v. bag, for example, with the solution of approximately 1 liter one would need to use lOOOx the amount of carrier protein/carrier protein- therapeutic agent and antibodies compared to that used in 1 mL. Thus, one cannot form the present nanoparticles in a standard i.v. bag. Furthermore, when the components are added to a standard i.v. bag in the therapeutic amounts of the present invention, the components do not self-assemble to form nanoparticles.
[0162] In one embodiment, the carrier protein or carrier protein-therapeutic agent particle is contacted with the binding agent in a solution having a pH between about 4 and about 8.
In one embodiment, the carrier protein or carrier protein-therapeutic agent particle is contacted with the binding agent in a solution having a pH of about 4. I n one embodiment, the carrier protein or carrier protein-therapeutic agent particle is contacted with the binding agent in a solution having a pH of about 5. In one embodiment, the carrier protein or carrier protein-therapeutic agent particle is contacted with the binding agent in a solution having a pH of about 6. In one embodiment, the carrier protein or carrier protein- therapeutic agent particle is contacted with the binding agent in a solution having a pH of about 7. In one embodiment, the carrier protein or carrier protein-therapeutic agent particle is contacted with the binding agent in a solution having a pH of about 8. In a preferred embodiment, the carrier protein or carrier protein-therapeutic agent particle is contacted with the binding agent in a solution having a pH between about 5 and about 7.
[0163] In one embodiment, the carrier protein particle or carrier protein-therapeutic agent particle is incubated with the binding agent at a temperature of about 5 °C to about 60 °C, or any range, subrange, or value within that range including endpoints. In a preferred embodiment, the carrier protein particle or carrier protein-therapeutic agent particle is incubated with the binding agent at a temperature of about 23 °C to about 60 °C.
[0164] Without being bound by theory, it is believed that the stability of the nanoparticles within the nanoparticle composition is, at least in part, dependent upon the temperature and/or pH at which the nanoparticles are formed, as well as the concentration of the components (i.e., carrier protein, binding agent, and optionally therapeutic agent) in the solution. In one embodiment, the Kd of the nanoparticles is between about 1 x 10"11 M and about 2 x 10"5M. In one embodiment, the Kd of the nanoparticles is between about 1 x 10"11 M and about 2 x 10"8 M. In one embodiment, the Kd of the nanoparticles is between about 1 x 10"11 M and about 7 x 10"9M. In a preferred embodiment, the Kd of the nanoparticles is between about 1 x 1 0 " 1 1 M and about 3x 10*M. Contemplated values include any value, subrange, or range within any of the recited ranges, including endpoints.
Lyophilization
[0165] The lyophilized compositions of this invention are prepared by standard
lyophilization techniques with or without the presence of stabilizers, buffers, etc.
Surprisingly, these conditions do not alter the relatively fragile structure of the
nanoparticles. Moreover, at best, these nanoparticles retain their size distribution upon
lyophilization and, more importantly, can be reconstituted for in vivo administration (e.g., intravenous delivery) in substantially the same form and ratios as if freshly made.
Formulations
[0166] In one aspect, the nanoparticle composition is formulated for systemic delivery, e.g., intravenous administration.
[0167] In one aspect, the nanoparticle composition is formulated for direct injection into a tumor. Direct injection includes injection into or proximal to a tumor site, perfusion into a tumor, and the like. Because the nanoparticle composition is not administered
systemically, a nanoparticle composition is formulated for direct injection into a tumor may comprise any average particle size. Without being bound by theory, it is believed that larger particles (e.g., greater than 500 nm, greater than 1 μπι, and the like) are more likely to be immobilized within the tumor, thereby providing what is believed to be a better beneficial effect.
[0168] In another aspect, provided herein is a composition comprising a compound provided herein, and at least one pharmaceutically acceptable excipient.
[0169] In general, the compounds provided herein can be formulated for administration to a patient by any of the accepted modes of administration. Various formulations and drug delivery systems are available in the art. See, e.g., Gennaro, A.R., ed. (1995) Remington's Pharmaceutical Sciences, 18th ed., Mack Publishing Co.
[0170] In general, compounds provided herein will be administered as pharmaceutical compositions by any one of the following routes: oral, systemic (e.g., transdermal, intranasal or by suppository), or parenteral (e.g., intramuscular, intravenous or
subcutaneous) administration.
[0171] The compositions are comprised of, in general, a compound of the present invention in combination with at least one pharmaceutically acceptable excipient.
Acceptable excipients are non-toxic, aid administration, and do not adversely affect the therapeutic benefit of the claimed compounds. Such excipient may be any solid, liquid, semi-solid or, in the case of an aerosol composition, gaseous excipient that is generally available to one of skill in the art.
[0172] Solid pharmaceutical excipients include starch, cellulose, talc, glucose, lactose, sucrose, gelatin, malt, rice, flour, chalk, silica gel, magnesium stearate, sodium stearate, glycerol monostearate, sodium chloride, dried skim milk and the like. Liquid and semisolid excipients may be selected from glycerol, propylene glycol, water, ethanol and various oils, including those of petroleum, animal, vegetable or synthetic origin, e.g., peanut oil, soybean oil, mineral oil, sesame oil, etc. Preferred liquid carriers, particularly for injectable solutions, include water, saline, aqueous dextrose, and glycols. Other suitable pharmaceutical excipients and their formulations are described in Remington's
Pharmaceutical Sciences, edited by E. W. Martin (Mack Publishing Company, 18th ed., 1990).
[0173] The present compositions may, if desired, be presented in a pack or dispenser device containing one or more unit dosage forms containing the active ingredient. Such a pack or device may, for example, comprise metal or plastic foil, such as a blister pack, or glass, and rubber stoppers such as in vials. The pack or dispenser device may be accompanied by instructions for administration. Compositions comprising a compound of the invention formulated in a compatible pharmaceutical carrier may also be prepared, placed in an appropriate container, and labeled for treatment of an indicated condition.
Treatment Methods
[0174] The nanoparticle compositions as described herein are useful in treating cancer cells and/or tumors in a mammal. In a preferred embodiment, the mammal is a human (i.e., a human patient). Preferably, the lyophilized nanoparticle composition is
reconstituted (suspended in an aqueous excipient) prior to administration.
[0175] In one aspect is provided a method for treating a cancer cell, the method
comprising contacting the cell with an effective amount of nanoparticle composition as described herein to treat the cancer cell. Treatment of a cancer cell includes, without limitation, reduction in proliferation, killing the cell, preventing metastasis of the cell, and the like.
[0176] In one aspect is provided a method for treating a tumor in a patient in need thereof, the method comprising administering to the patient a therapeutically effective amount of a nanoparticle composition as described herein to treat the tumor. In one embodiment, the
size of the tumor is reduced. In one embodiment, the tumor size does not increase (i.e. progress) for at least a period of time during and/or after treatment.
[0177] In one embodiment, the nanoparticle composition is administered intravenously. In one embodiment, the nanoparticle composition is administered directly to the tumor. In one embodiment, the nanoparticle composition is administered by direct injection or perfusion into the tumor.
[0178] In one embodiment, the method comprises: a) administering the nanoparticle composition once a week for three weeks; b) ceasing administration of the nanoparticle composition for one week; and c) optionally repeating steps a) and b) as necessary to treat the tumor.
[0179] In one embodiment, the therapeutically effective amount of the nanoparticles described herein comprises about 50 mg/m2 to about 200 mg/m2 carrier protein or carrier protein and therapeutic agent. In a preferred embodiment, the therapeutically effective amount comprises about 75 mg/m2 to about 175 mg/m2 carrier protein or carrier protein and therapeutic agent. Contemplated values include any value, subrange, or range within any of the recited ranges, including endpoints.
[0180] In one embodiment, the therapeutically effective amount comprises about 20 mg/m2 to about 90 mg/m2 binding agent, e.g., antibody, aptamer or Fc fusion. In a preferred embodiment, the therapeutically effective amount comprises 30 mg/m2 to about 70 mg/m2 binding agent, e.g., antibody, aptamer or Fc fusion. Contemplated values include any value, subrange, or range within any of the recited ranges, including endpoints.
[0181] Cancers or tumors that can be treated by the compositions and methods described herein include, but are not limited to: biliary tract cancer; brain cancer, including glioblastomas and medulloblastomas; breast cancer; cervical cancer; choriocarcinoma; colon cancer; endometrial cancer; esophageal cancer, gastric cancer; hematological neoplasms, including acute lymphocytic and myelogenous leukemia; multiple myeloma; AIDS associated leukemias and adult T-cell leukemia lymphoma; intraepithelial neoplasms, including Bowen's disease and Paget's disease; liver cancer (hepatocarcinoma); lung cancer; lymphomas, including Hodgkin's disease and lymphocytic lymphomas;
neuroblastomas; oral cancer, including squamous cell carcinoma; ovarian cancer, including those arising from epithelial cells, stromal cells, germ cells and mesenchymal cells;
pancreas cancer; prostate cancer; rectal cancer; sarcomas, including leiomyosarcoma, rhabdomyosarcoma, liposarcoma, fibrosarcoma and osteosarcoma; skin cancer, including melanoma, Kaposi's sarcoma, basocellular cancer and squamous cell cancer; testicular cancer, including germinal tumors (seminoma, non-seminoma[teratomas,
choriocarcinomas]), stromal tumors and germ cell tumors; thyroid cancer, including thyroid adenocarcinoma and medullar carcinoma; and renal cancer including adenocarcinoma and Wilms tumor. In important embodiments, cancers or tumors include breast cancer, lymphoma, multiple myeloma, and melanoma.
[0182] In general, the compounds of this invention will be administered in a
therapeutically effective amount by any of the accepted modes of administration for agents that serve similar utilities. The actual amount of the compound of this invention, i.e., the nanoparticles, will depend upon numerous factors such as the severity of the disease to be treated, the age and relative health of the subject, the potency of the compound used, the route and form of administration, and other factors well known to the skilled artisan.
[0183] An effective amount of such agents can readily be determined by routine experimentation, as can the most effective and convenient route of administration, and the most appropriate formulation. Various formulations and drug delivery systems are available in the art. See, e.g., Gennaro, A.R., ed. (1995) Remington's Pharmaceutical Sciences, 18th ed., Mack Publishing Co.
[0184] An effective amount or a therapeutically effective amount or dose of an agent, e.g., a compound of the invention, refers to that amount of the agent or compound that results in amelioration of symptoms or a prolongation of survival in a subject. Toxicity and therapeutic efficacy of such molecules can be determined by standard pharmaceutical procedures in cell cultures or experimental animals, e.g., by determining the LD50 (the dose lethal to 50% of the population) and the ED50 (the dose therapeutically effective in 50% of the population). The dose ratio of toxic to therapeutic effects is the therapeutic index, which can be expressed as the ratio LD50/ED50. Agents that exhibit high therapeutic indices are preferred.
[0185] The effective amount or therapeutically effective amount is the amount of the compound or pharmaceutical composition that will elicit the biological or medical response of a tissue, system, animal or human that is being sought by the researcher, veterinarian, medical doctor or other clinician. Dosages may vary within this range depending upon the dosage form employed and/or the route of administration utilized. The exact formulation, route of administration, dosage, and dosage interval should be chosen according to methods known in the art, in view of the specifics of a subject's condition.
[0186] Dosage amount and interval may be adjusted individually to provide plasma levels of the active moiety that are sufficient to achieve the desired effects; i.e., the minimal effective concentration (MEC). The MEC will vary for each compound but can be estimated from, for example, in vitro data and animal experiments. Dosages necessary to achieve the MEC will depend on individual characteristics and route of administration. In cases of local administration or selective uptake, the effective local concentration of the drug may not be related to plasma concentration.
EXAMPLES
[0187] The present disclosure is illustrated using nanoparticles composed of albumin- bound paclitaxel (i.e., ABRAXA E®) or cisplatin as core, and bevacizumab (i.e., Avastin®) or Rituximab (i.e., Rituxan®) as antibodies.
[0188] One skilled in the art would understand that making and using the nanoparticles of the Examples are for the sole purpose of illustration, and that the present disclosure is not limited by this illustration.
[0189] Any abbreviation used herein, has normal scientific meaning. All temperatures are °C unless otherwise stated. Herein, the following terms have the following meanings unless otherwise defined:

BEV — bevacizumab
BSA bovine serum albumin dH20 — distilled water
DM EM Dulbecco's Modified Eagle's
Medium nM — nanomolar
EdU — 5-ethynyl-2'-deoxyuridine
EM electron microscopy
FCB — flow cytometry buffer
FITC — Fluorescein
1<D kilo-dalton
Kd — dissociation constant kg — kilogram
KV kilo-volts
L/hr — liter/hour
LC-MS liquid chromatography-mass spectrometry
M molar mCi — millicuries mg milligram
ml or mL — milliliter m2 — square meters mm3 cubic millimeter
— microgram μΐ — microliter μπι mi crometer/mi cron
PBS — Phosphate buffered saline pK — pharmacokinetics
RT room temperate φηι — rotations per minute
V — volts x g times gravity
Example 1: Nanoparticle Preparation
[0190] ABRAXA E® (ABX) (10 mg) was suspended in bevacizumab (BEV) (4 mg [160 μΐ] unless otherwise indicated), and 840 μΐ of 0.9% saline was added to give a final concentration of 10 mg/ml and 2 mg/ml of ABX and BEV, respectively. The mixture was incubated for 30 minutes at room temperature (or at the temperature indicated) to allow particle formation. For Mastersizer experiments to measure particle size of ABX:BEV complexes, 10 mg of ABX was suspended in BEV at concentrations of 0 to 25 mg/ml. Complexes of ABX with rituximab (0-10 mg/ml) or trastuzumab (0-22 mg/ml) were formed under similar conditions.
[0191] For use in humans, the ABX:BEV complexes may be prepared by obtaining the dose appropriate number of 4 mL vials of 25 mg/mL BEV and diluting each vial per the following directions to 4 mg/mL. The dose appropriate number of 100 mg vials of ABX can be
prepared by reconstituting to a final concentration containing 10 mg/mL ABX nanoparticles. Using a sterile 3 mL syringe, 1.6 mL (40 mg) of bevacizumab (25 mg/mL) can be withdrawn and slowly injected, over a minimum of 1 minute, onto the inside wall of each of the vials containing 100 mg of ABX. The bevacizumab solution should not be injected directly onto the lyophilized cake as this will result in foaming. Then, using a sterile 12 mL sterile syringe, 8.4 mL 0.9% Sodium Chloride Injection, USP, can be withdrawn and slowly injected, over a minimum of 1 minute, 8.4 mL onto the inside wall of each vial containing ABX 100 mg and BEV 40 mg. Once the addition of BEV 1.6 mL and 0.9% Sodium Chloride Injection, USP 8.4 mL is completed, each vial can be gently swirled and/or inverted slowly for at least 2 minutes until complete dissolution of any cake/powder occurs. Generation of foam should be avoided. At this point, the concentration of each vial should be 100 mg/10 mL ABX and 40 mg/10 mL BEV. The vials containing the ABX and BEV should sit for 60 minutes. The vial(s) should be gently swirled and/or inverted every 10 minutes to continue to mix the complex. After 60 minutes has elapsed, the calculated dosing volume of ABX and BEV should be withdrawn from each vial and slowly added to an empty viaflex bag. An equal volume of 0.9% Sodium Chloride Injection, USP is then added to make the final concentration of ABX 5 mg/mL and BEV 2 mg/mL. The bag should then be gently swirled and/or inverted slowly for 1 minute to mix. The ABX:BEV nanoparticles can be stored for up to 4 hours at room temperature following final dilution.
Example 2: Binding of ABX and BEV in vitro
[0192] To determine whether ABX and BEV interact, the nanoparticles formed in Example 1 were analyzed by flow cytometry and electron microscopy.
Methods
[0193] Flow Cytometry: AB160 was produced as described in Example 1 above. To determine binding of BEV to ABX, visualization of AB 160 was performed on an Accuri C6 flow cytometer (BD Franklin Lakes, NJ) and data analysis was done using Accuri C6 software. Biotinylated ^g) goat anti-mouse IgG (Abeam, Cambridge, MA) was labeled with 5 μg of streptavidin PE (Abeam, Cambridge, MA). The goat anti-mouse IgG was chosen to label AB160 because the Fab portion of the BEV is mouse derived. ABX and AB160 were incubated with the PE-labeled goat anti-mouse IgG for 30 minutes at room temperature, washed and visualized by flow cytometry.
[0194] Electron Microscopy: Five μΐ ABX, dissolved in PBS at 6 mg/ml, was added to a 300-mesh parlodian-carbon coated copper grid and allowed to sit for 1 minute. A pointed piece of filter paper was touched to the drop to remove excess liquid, leaving a thin film on the grid. The grids were allowed to dry. To dissolve the buffer crystals left on the dried grid, the sample was washed three times in dH20. A small drop of 1% phosphotungstic acid (PTA), pH 7.2, was added to the grid. The grid was then again touched by a pointed piece of filter paper to remove excess liquid, leaving a thin film on the grid and allowed to dry. BEV (Genentech) at 25 mg/ml in 0.9% sodium chloride solution was diluted with PBS at 1 : 10 ratio. Five μΐ of BEV was loaded on nickel formvar-coated grid and allowed to air dry for 30 minutes to 1 hour. For the AB160, 10 mg/ml ABX, dissolved in PBS, and 4mg/ml BEV, in 0.9% sodium chloride solution, were mixed at 2.5: 1 ratio. The complex was further diluted with PBS at 1 :5. Five μΐ of the complex was loaded on nickel formvar-coated grid and air dried for 30 minutes to 1 hour. Both samples were incubated for 1 hour in goat anti-mouse IgG with 6 nm gold- conjugated particles (Electron Microscopy Sciences), diluted 1 :30 with 10% FCB/PBS, washed 6 times with PBS (each 2 minutes), 6 times with dH20, then stained with the mixture of 2% methylcellulose and 4% UA (9: 1) for 5 minutes. Filter paper was used to drain the stain and the grid was air dried for 1 hour. Both samples were incubated overnight in donkey anti-mouse IgG with 6 nm gold-conjugated particles (Jackson ImmunoResearch) diluted 1 :25 with 10% FCB/PBS, washed 6 times with PBS (each 2 minutes), 6 times with dH20 water, stained with 1% PTA for 5 minutes, air dried, covered with 2% methylcellulose, and air dried for 1 hour. The micrographs were taken on a JEOL1400 at operating at 80 KV.
Results
[0195] ABX (10 mg/ml) was co-incubated with 4 mg/ml BEV in vitro and found that they formed 1 60 nm nanoparticles (referred to herein as AB 160). Because the Fab portion of the IgGI (BEV) is of mouse origin, particles containing BEV were selectively labeled with purified goat anti-mouse IgG followed by anti-goat PE as a secondary antibody. As a negative control, samples were stained with the anti-goat PE only.
Particles were visualized by flow cytometry and demonstrated a bright signal of anti- mouse IgGI binding to AB160 (41.2% positive) relative to ABX (6.7% positive) alone (FIG. IA). To validate binding of BEV to ABX, the BEV were labeled with gold- labeled mouse anti-human IgG and the particles were visualized with electron microscopy
(FIG IB). Surprisingly, the EM pictures suggest a monolayer of BEV surrounding ABX nanoparticles.
[0196] To determine what protein (albumin or BEV) the paclitaxel remains bound to when the complex breaks down, AB 160 were made and collected fractions: the particulate (nanoAB160), proteins greater than 100 kD and proteins less than 100 kD. Paclitaxel was measured in each fraction by liquid chromatography-mass spectrometry (LC-MS). Roughly 75% of the paclitaxel remained within the particulate, and the majority of the remaining paclitaxel was associated with the fraction containing proteins 100 kD or greater (FIG. 1 C, top), suggesting that when the particulate dissociates the paclitaxel is bound to BEV alone or a BEV and albumin heterodimer. This indicates that the dissociated complexes contain the chemotherapy drug with the antibody, which would still traffic to the high-VEGF tumor microenvironment. These findings were confirmed by Western blot analysis of the supernatants from AB160, which showed that BEV and paclitaxel co-localize at approximately 200 kD, a size consistent with a paclitaxel-BEV- albumin protein complex (FIG. I C, bottom).
Example 3: Function of AB160 in vitro
[0197] Confirmation that the two key elements in the complexes, the antibody and the paclitaxel, retained their function when present in the complexes was demonstrated.
Methods
[0198] In vitro toxicity: The A375 human melanoma cell line (ATCC Manassas, VA) and Daudi B-cell lymphoma line (ATCC Manassas, VA) were cultured in DMEM with 1%
6
PSG and 10% FBS. Cells were harvested and plated at 0.75 x 10 cells per well in 24 well plates. Cells were exposed to ABX or AB160 at paclitaxel concentrations from 0 to 200 μg/ml overnight at 37 °C and 5% C02. To measure proliferation, the Click-iT EdU (Molecular Probes, Eugene, OR) kit was utilized. Briefly, 10 mM EdU was added to the wells and incubated overnight with the cells and ABX or AB 160. The cells were permeabilized with 1% saponin and intercalated EdU was labeled with a FITC-conjugated antibody. The proliferation index was determined by dividing the FITC positive cells from each treatment by the maximum proliferation of untreated EdU labeled cells.
[0199] VEGF ELISA: To determine whether BEV can still bind its ligand, VEGF, when bound to ABX, a standard VEGF ELISA (R and D Systems, Minneapolis, MN) was employed. AB 160 was prepared as described and 2000 pg/ml VEGF was added to the AB160 complex or ABX alone. The VEGF was incubated with the nanoparticles for 2 hours at room temperature. The suspension was spun at 6000 rpm for 15 minutes, supernatants were collected and free VEGF was measured by ELISA. Briefly, ELISA plates were coated with capture antibody overnight at 4 °C. Plates were washed, blocked and standards and samples were added. After washing, detection antibody was added and plates were developed with substrate (R and D Systems, Minneapolis, MN). Absorbance was measured at 450 nm using a Versamax ELISA plate reader (Molecular Devices, Sunnyvale, CA). The concentration of unbound VEGF was determined with a standard curve from 0 to 2000 pg/ml.
Results
[0200] AB160 has similar toxicity relative to ABX alone in an in vitro toxicity assay with the human melanoma cell line, A375, suggesting that the paclitaxel functions equally in either formulation (FIG. 1 D).
[0201] To test the binding of VEGF to BEV in the AB160 complex, AB 160 or ABX was co- incubated with VEGF, the particulate removed, and the supernatant tested for VEGF content. The lack of VEGF in the supernatant measured from AB160 (<10% VEGF unbound) indicated that the VEGF was bound by the BEV in the AB 160 complex, while it remained free when incubated with the ABX (>80% VEGF unbound) alone (FIG. 1 E).
[0202] Importantly, these assays demonstrated that the paclitaxel in AB 160 retains its toxicity to tumor cells and the bound BEV maintains the ability to bind its ligand, VEGF.
Example 4: Particle Size and Protein Affinity
[0203] To understand the characteristics of the nanoparticles formed when binding BEV to ABX, the size of the ABX:BEV complexes was determined relative to ABX.
Methods
[0204] Mastersizer and Nanosight: The particle size of ABX and antibody-ABX drug complexes were measured by dynamic light scattering on a Mastersizer 2000 (Malvern Instruments, Westborough, MA). To measure particle size, 2 ml (5 mg/ml) of
ABRAXA E® or complex was added to the sample chamber. Data were analyzed with Malvern software and particle size distributions were displayed by volume. The particle sizes and stability were later validated using the Nanosight System (Malvern Instruments, Westborough, MA). The ABX or complex particles were diluted to the appropriate range to accurately measure particle sizes. Data was displayed by particle size distribution;
however, the nanoparticle tracking analysis uses Brownian motion to determine particle size.
[0205] Binding Assay: Biotinylated BEV, rituximab or trastuzumab at 100 μg/ml was bound to the streptavidin probe (ForteBio Corp. MenloPark, CA). The binding of ABX was measured by light absorbance on the BLitz system (ForteBio Corp. MenloPark, CA) at 1000, 500 and 100 mg/ml. The association and dissociation constants were calculated using the BLitz software.
[0206] Bio-Layer Interferometry (BLitz) technology was utilized to assess the binding affinity of BEV to ABX. Biotinylated BEV was bound to the streptavidin probe and exposed to ABX (1000, 500, and 100 μg/ml). The dissociation constant (Kd) of BEV and
8
ABX is 2.2 x 10" M at room temperature and pH 7, consistent with a strong non-covalent interaction. The binding affinity of BEV and ABX is within the range of dissociation constants observed between albumin and natural or engineered albumin-binding domains of some bacterial proteins. Nilvebrant, J. et al. (2013) Comput Struct Biotechnol J
6:e201303009.
Results
[0207] ABX:BEV nanoparticles were consistently larger (approximately 160 nm) than the 130 nm ABX alone (Fig 2 A). The size of the nanoparticle created directly correlated to the concentration of BEV used, with median sizes ranging from 0.157 to 2.166 μπι. (FIG. 2A). With the goal of these studies being a Phase I clinical trial, the smallest ABX:BEV particle (AB160) were focused on because it is the most similar to the 130 nm ABX. The size of the AB160 particle was consistent with ABX plus a monolayer of BEV surrounding it and with the EM image of the particle (see FIG. IB).
[0208] To determine whether intravenous administration conditions affect nanoparticle size distributions, the particle size distributions of AB160 (or ABX) incubated in saline for up to 24 hours at room temperature were evaluated. AB 160 size distribution does not
significantly change for up to 24 hours (FIGs. 9A and 9B). However, by 4 hours at room temperature, there is some evidence of AB160 breakdown by ELISA (FIG. 9C).
[0209] To determine the stability of AB160 in plasma, ABX or AB160 was incubated in saline or heparinized human plasma at relative volume ratios of 9: 1 or 1 : 1. Notably, no particles (0.01 to 1 μπι^εΓε detected when either ABX (FIG. 10, top panel) or AB 160 (FIG. 10, bottom panel) is incubated in plasma at equal volumes (1 : 1).
[0210] Western blot (data not shown) indicated that, in saline or heparinized human plasma, the AB 160 dissociated into smaller protein conjugates that still contain the tumor- targeting antibody, albumin and the cytotoxic agent, paclitaxel. These protein conjugates retain their ability to target the tumor and, once at the tumor site, can quickly dissolve and release the cytotoxic payload to effectively initiate tumor regression without internalization of the entire nanoparticle by tumor cells.
[0211] Next, the ABX was suspended in BEV and the mixture diluted with saline at pH 3, 5, 7, or 9 prior to incubation at various temperatures (RT, 37 °C and 58 °C) to allow particle formation in order to test whether binding affinity was pH- and/or temperature- dependent. The binding affinity of ABX and BEV is both pH- and temperature- dependent, with the highest binding affinity observed when the particles are formed at pH 5 and 58 °C (FIG. 2B).
[0212] To determine if the higher affinity binding of BEV and ABX at 58 °C and pH 5 translated into stability of the complex, various preparations were compared by
nanoparticle tracking analysis (Nanosight). The stability of AB160 prepared at 58 °C and pH 5 (AB 1600558), room temperature and pH 7 (AB 16007), or 58 °C and pH 7
(AB1600758) was compared to ABX exposed to the same conditions (ABX0558, ABX07, and ABX0758, respectively) after incubation in human AB serum for 0, 15, 30, or 60 minutes.
[0213] The particles made under higher affinity conditions (pH 7 and 58 °C) were also more stable, as indicated by the number of particles present per mg ABX after exposure to human AB serum. The AB160 particles exhibited increased stability in human serum that correlated with their binding affinities. In particular, AB16007 and AB 1600558 were more stable in both saline and human serum than ABX alone, as determined by size and number of particles measured per mg ABX (FIG. 2C and Table 3). This shows that the
stability of AB160 particles can be manipulated by changing the conditions under which the AB 160 particles are formed.
Table 3: Stability of AB160 and ABX in human AB serum

[0214] Particles per mg ABX x 10
[0215] [0203] These data demonstrated that BEV binds to ABX with affinity in the picomolar range, indicating a strong non-covalent bond, and demonstrated a particle size distribution consistent with ABX surrounded by a monolayer of antibody molecules; the size of the particles created is dependent on the antibody concentration.
Example 5: Efficacy of AB160 in Mice
[0216] A xenograft model of A375 human melanoma cells implanted into athymic nude mice was employed to test AB160 efficacy in vivo.
Methods
[0217] In vivo experiments were performed at least 2 times. The number of mice required for those experiments was determined by power analysis. Mouse tumors were measured 2-3 times/week and mice were sacrificed when the tumor was 10% by weight. Mice that had complete tumor responses were monitored for 60-80 days post-treatment. The end point of the mouse studies was median survival. Kaplan-Meier curves were generated and Mantle-Cox test was performed to determine significance of median survival between treatment groups. The in vitro results presented are representative of at least 5 repeated experiments. Statistical analyses of in vitro and in vivo percent change from baseline experiments were done using the Student's t-test.
[0218] Mouse Model: To test tumor efficacy, 1 x 106 A375 human melanoma cells were implanted into the right flank of athymic nude mice (Harlan Sprague Dawley,
Indianapolis, IN). When the tumors had reached a size of about 700 mm3, the mice were randomized and treated with PBS, ABX (30 mg/kg), BEV (12 mg/kg), BEV followed by ABX, or AB 160 at the above concentrations. For the mouse experiments testing bigger AB particles, the highest dose of BEV (45 mg/kg) necessary to create the larger particles was used in the BEV-only treatment group. Tumor size was monitored 3 times/week and tumor volume was calculated with the following equation: (length x width2)/2. Mice were sacrificed when the tumor size equaled 1 0% of the mouse body weight or about 2500 mm3. The day 7 percent change from baseline was calculated as follows: [(tumor size on treatment day -tumor size on day 7)/tumor size on treatment day] x 100. The in vivo
6
testing of the AR160 was similar except 5x10 Daudi cells were injected into the right flank of athymic nude mice.
Results
[0219] AB160 was tested relative to PBS, the single drugs alone, and the drugs administered sequentially. Mice treated with AB 160 had significantly reduced tumor size compared to all other treatment groups (p=0.0001 to 0.0089) at day 7 post-treatment, relative to baseline (FIG. 3 A). Tumors in all of the mice treated with AB 160 had regressed at day 7, and this tumor response translated into significantly increased median survival of the AB 160 group relative to all other groups (FIG. 3B), with a survival of 7, 14, 14, 18 and 33 days for the PBS (pO.0001), BEV (p=0.003), ABX (p=0.0003), BEV + ABX (p=0.0006) and AB160 groups, respectively.
[0220] It is likely that larger tumors have a higher local VEGF concentration. When data were analyzed based on the size of the tumor on day of treatment (<700mm3 and
>700mm3), the larger tumors were shown to have a greater response to AB 160, suggesting that higher tumor VEGF concentration attracts more BEV-targeted ABX to the tumor. The difference in the percent change from baseline was significant for the AB 160 groups (p=0.0057). This observation was not seen in the ABX only (p=0.752) group, where the ABX has no targeting capability (FIG. 3C).
[0221] Particles of increasing size were prepared using increasing BEV:ABX ratios as shown in FIG. 2A. Tumor regression and median survival positively correlated with
increasing particle size, indicating that biodistribution of larger particles may be altered relative to the smaller ones (FIGs. 3D and 3E). Full toxicity studies were performed on the mice and no toxicities were noted.
Example 6: Paclitaxel Pharmakokinetics in Mice
[0222] To compare the pharmacokinetics (pk) of AB 160 and ABX, plasma paclitaxel concentrations were measured in mice administered AB160 or ABX at 0, 4, 8, 12 and 24 hours.
Methods
[0223] Paclitaxel Pharmacokinetics: The liquid chromatographic separation of paclitaxel and d5 paclitaxel were accomplished using an Agilent Poroshell 120 EC-C18 precolumn (2.1 x 5 mm, 2.7 μιη, Chrom Tech, Apple Valley, MN) attached to an Agilent Poroshell 120 EC-C18 analytical column (2.1 x 100 mm, 2.7 μιη Chrom Tech, Apple Valley, MN) at 40 °C, eluted with a gradient mobile phase composed of water with 0.1% formic acid (A) and ACN with 0.1% formic acid (B) with a constant flow rate of 0.5 ml/minute. The elution was initiated at 60% A and 40% B for 0.5 minutes, then B was linearly increased from 40-85% for 4.5 minutes, held at 85% B for 0.2 minutes, and returned to initial conditions for 1.3 minutes. Autosampler temperature was 10 °C and sample injection volume was 2 μΐ. Detection of paclitaxel and the internal standard d5-paclitaxel were accomplished using the mass spectrometer in positive ESI mode with capillary voltage 1.75 kV, source temp 150 °C, desolvation temp 500 °C, cone gas flow 150 L/hr, desolvation gas flow 1000 L/hr, using multiple reaction monitoring (MRM) scan mode with a dwell time of 0.075 seconds. The cone voltages and collision energies were determined by MassLynx-Intelli start, v4.1, software and varied between 6-16 V and 12-60 eV, respectively. The MRM precursor and product ions were monitored at m/z 854.3> 105.2 for paclitaxel and 859.3>291.2 for d5 paclitaxel. The primary stock solutions of paclitaxel (1 mg/ml in EtOH) and d5 paclitaxel (1 mg/ml in EtOH) were prepared in 4 ml amber silanized glass vials and stored at -20 °C. Working standards were prepared by dilution of the stock solution with ACN in 2 ml amber silanized glass vials and stored at -20 °C. Plasma samples were extracted as follows, 100 μΐ plasma sample was added to a 1.7 ml microcentrifuge tube containing d5 paclitaxel (116.4 nM or 100 ng/ml) and 300 μΐ ACN, vortexed, incubated at room temperature for 10 minutes to precipitate proteins, and centrifuged (14,000 rpm) or 3 minutes. The supernatant was filtered on an Agilent
Captiva NDlipids plate (Chrom Tech, Apple Valley, MN), collected in a deep 96-well plate, and dried using nitrogen gas. The samples were reconstituted using 100 μΐ ACN and shaken on a plate shaker (high speed) for 5 minutes. Plasma standard curves were prepared daily containing paclitaxel (0.59-5855 nM or 0.5-5000 ng/ml) and d5 paclitaxel (116.4 nM) for paclitaxel quantitation. Mouse tumors were thawed on ice, weighed, and diluted 2 parts (weight to volume) in l x PBS. Tumors were then homogenized using a PRO200 tissue homogenizer using the saw tooth probe (5 mm x 75 mm). Tumor homogenate was than processed the same as the human plasma samples.
[0224] Mouse Imaging: Avastin and IgG control solutions were prepared and 1 -125 labeled per protocol (Imanis Life Sciences). Briefly, Tris Buffer (0.125 M Tris-HCl, pH
6.8, 0.15 M NaCl) and 5 mCi Na125 I were added directly to iodination tubes
(ThermoFischer Scientific, Waltham, MA). The iodide was allowed to activate and was swirled at room temperature. Activated iodide was mixed with the protein solution. 50 μΐ of Scavenging Buffer (10 mg tyrosine/mL in PBS, pH 7.4) was added and incubated for five minutes. After addition of Tris/BSA buffer and mixing, samples were applied in 1 O K MWCO dialysis cassettes against pre-cooled PBS for 30 minutes, 1 hour, 2 hours, and overnight at 4 °C. Radioactivity was determined by Gamma counter, then
disintegrations per minute (DPM) and specific activity were calculated. Mice were injected in their tail vein with Avastin 1-125, ABRAXA E®-A VASTEST® 1-125,
ABRAXA E®-human IgG 1-125, or ABRAXANE® only. Animals were imaged at 3, 10, 24 and 72 hours post-administration via SPECT- CT imaging using the U-SPECT- II/CT scanner (MILabs, Utrecht, The Netherlands). SPECT reconstruction was performed using a POSEM (pixelated ordered subsets by expectation maximization) algorithm. CT data were reconstructed during the Feldkamp algorithm. Co-registered images were further rendered and visualized using PMOD software (PMOD Technologies, Zurich, Switzerland). Animals were sacrificed and dissected at 72 hours post- injection. Selected tissues and organs of interest were measured using radioisotope dose calibrator (Capintec CRC-127R, Capintec Inc.).
Results
[0225] Results of the first pk experiment are provided in FIGs. 4A and 4B. The area under the curve (AUC) and maximum serum concentration (Cmax) were calculated in A375 tumor bearing and non-tumor bearing mice. In the first pk experiment the Cmax and
AUC were very similar in the non-tumor bearing mice for AB160 and ABX (63.3+Λ39.4 vs. 65.5+/-14.4 and 129 vs. 133 μg/ml, respectively). However, in the tumor bearing mice, the Cmax and AUC for the treatment groups were different (55.7+/-21.2 vs 63.3+/- 17.3 and 112 vs 128 μg/ml, respectively) (FIG. 4C). Although this difference was not statistically significant, it is consistent with superior targeting by AB 160, relative to ABX.
[0226] A second pk experiment was performed with additional early time points and large versus small tumor sizes (FIGs. 4D-4F). The results of this experiment demonstrated smaller AUC in tumor bearing mice relative to non-tumor bearing mice, with the lowest blood values of paclitaxel in the large tumor mice relative to the small tumor mice (80.4+/-2.7, 48.4+/-12.3, and 30.7+/-5.2 for ABX-treated non-tumor, small tumor and large tumor bearing mice, respectively; 66.1+/-19.8, 44.4+/-12.1 and 22.8+/-6.9 for AB160-treated). Similarly, the CmaX dropped in both treatment groups in mice with larger tumors (47.2, 28.9 and 19.7 μg/ml for ABX and 40.1, 26.9 and 15.3 μg/ml for AB 160) (FIG. 4G). The AUC and Cmax of paclitaxel in blood were lower in AB160-treated mice relative to ABX-treated mice. Although not statistically significant, this data is further consistent with higher deposition of paclitaxel in the tumors treated with AB 160.
[0227] To directly test this hypothesis, tumor paclitaxel concentrations by LC-MS were measured. The tumor paclitaxel concentration was significantly higher in tumors treated with AB160 relative to ABX at the 4 hour (3473 μg/mg of tissue +/-340 vs 2127 μg/mg of tissue +/- 3.5; p=0.02) and 8 hour (3005 μg/mg of tissue +/- 146 vs 1688 μg/mg of tissue +/- 146; p=0.01) time points, suggesting paclitaxel stays in the tumor longer when targeted by the antibody (FIG. 4H). This explains the blood pk and is consistent with redistribution of drug to tissues including the tumor.
[0228] Live in vivo imaging of 1-125 labeled AB160 (Abx-AvtI125) and IgG isotype bound ABX (Abx-IgGI125) confirmed the results of the LC-MS, with higher levels of I- 125 in the tumor of mice treated with AB160 relative to IgG-ABX at 3 hours (32.2 uCi/g +/- 9.1 vs 18.5 uCi/g +/- 1.65; p=0.06) and 10 hours (41.5 uCi/g +/- 6.4 vs 28.7 uCi/g +/- 2.66; p=0.03) post injection (FIGs. 41 and 4J). Taken together, these data demonstrate that binding BEV to ABX alters blood pk, and this alteration is due to a redistribution of the drug to the tumor tissue as shown by both LC-MS of paclitaxel and 1-125 labeling of BEV relative to an isotype matched IgGl.
[0229] Without being bound by theory, it is believed that by binding a tumor-targeted antibody to ABX, the pk is altered more dramatically than ABX alone, lowering the Cmax and AUC in the blood because of redistribution of AB160 to the tumor tissue. These results from mouse blood paclitaxel pk, tumor tissue levels of paclitaxel, and 1-125 radioactivity levels in mice treated with AB160 relative to ABX alone suggest that antibody targeting of the ABX alters biodistribution of paclitaxel such that increased levels reach the tumor and are retained there for a longer period of time, yielding enhanced tumor regression.
Example 7: Binding of Other Therapeutic Antibodies
[0230] The binding of the anti-human CD20 antibody (rituximab) and the anti-HER2/neu receptor antibody (trastuzumab) to ABX was tested to determine if other IgG therapeutic antibodies also exhibit binding to ABX when combined ex vivo.
Methods
[0231] Nanoparticles containing rituximab or trastuzumab were prepared and tested as described in the above examples.
Results
[0232] The particle size of the complexes with both BEV and trastuzumab (HER) were very similar, with average sizes ranging from 0.157 to 2.166 μιη (FIG. 2A) and 0.148 to 2.868 μπι (FIG. 5B), respectively. In contrast, particles formed with rituximab became much larger at lower antibody: ABX ratios, with particle sizes ranging from 0.159 to 8.286 μιη (FIG. 5A).
[0233] The binding affinities of rituximab and trastuzumab with ABX were determined by BLitz under variable pH. Both antibodies bind with relatively high affinity in the picomolar range (FIG. 5C). The rituximab affinity to ABX decreased with higher pH, but trastuzumab affinity to ABX was unaffected by pH (FIG. 5C).
[0234] The efficacy of the 160 nm particle made with rituximab (AR160) was tested in vitro and in vivo. In vitro, the B-cell lymphoma cell line Daudi was treated with AR160, ABX, or rituximab alone at increasing concentrations (0 to 200 μg/ml) of paclitaxel. AR160 (IC50=10μg/ml) significantly inhibited proliferation of Daudi cells treated for 24
hours (p=0.024) compared to either ABX (IC50>20(^g/ml) or rituximab (IC50>20(^g/ml) alone (FIG. 6A).
[0235] In vivo, a xenotransplant model of Daudi cells was established in athymic nude mice. Once tumors were established, mice were treated with PBS, ABX, rituximab, ABX and rituximab given sequentially, or AR160. On day 7 post treatment, tumors were measured and the percent change in tumor size from baseline was calculated. AR160- treated tumors regressed or remained stable, while tumors in all other treatment groups progressed (FIG. 6B). The percent change from baseline tumor size in the AR160 group compared to all other groups was significant (p<0.0001). The mice treated with AR160 had a significantly longer median survival of greater than 60 days compared to 12, 16, and 12 days for mice treated with PBS (p<0.0001), ABX (p<0.0001), or rituximab (p=0.0002), respectively (FIG. 6C). However, the difference in median survival was not significant between AR160 and the sequentially treated groups (p=0.36). This may be because the rituximab binds to the tumor cells and remains on the cell surface, allowing the subsequently-administered ABX to bind to the antibody when it enters the tumor site, unlike BEV which binds a soluble target and not a cell surface marker.
Example 8: Binding of Other Chemotherapy Drugs to AB160
[0236] The efficacy of other chemotherapy drugs to form functional nanoparticles was evaluated.
Methods
[0237] Nanoparticles containing cisplatin were prepared and tested as described in the above examples.
Results
[0238] To test if another chemotherapy drug could bind to the AB 160 particles, cisplatin and ABX were co-incubated and the amount of free cisplatin remaining in the supernatant was measured by HPLC. Approximately 60% (i.e., only 40% remains in the supernatant) of the cisplatin bound to the ABX (FIG. 7A).
[0239] Next, tumor toxicity of AC relative to ABX and cisplatin alone was tested using A375 cells. The complexes were centrifuged to remove highly toxic unbound cisplatin, and reconstituted in media to ensure that any additional toxicity of AC relative to ABX is
due only to ABX-bound cisplatin. For parity, the ABX only was centrifuged in a similar manner. AC (IC50=9C^g/ml) inhibited proliferation of A375 cells to a greater extent than ABX alone (IC50> 100C^g/ml) (FIG. 7B). The diminished toxicity in this experiment relative to other toxicity experiments is due to some loss of drug in the centrifugation step, but the comparison of ABX to AC remains relevant.
[0240] To determine the tumor toxicity of cisplatin-containing AB160 complexes, AB 160 was co-incubated with cisplatin to form cisplatin containing particles (ABC complex). The ABC complex was tested in the A375 melanoma xenotransplant model relative to each drug alone and AB 160. Tumors treated with AB160, AB160 + cisplatin given
sequentially, and the ABC complex all showed regression in tumor size at 7 days post treatment (FIG. 7C), but the ABC complex conferred the longest median survival (35 days, relative to AB160 and AB 160 + cisplatin at 24 and 26 days, respectively). Although the difference was not statistically significant (p= 0.82 and 0.79) (FIG. 7D), the data is consistent with benefits of the ABC complex to long- term survival rates.
[0241] These data demonstrated that the albumin portion of the ABX provides a platform for other therapeutic antibodies to bind, such as rituximab and trastuzumab, as well as other chemotherapy agents (e.g., cisplatin), which all had similar efficacy in vitro and in vivo as AB160.
[0242] Together these data demonstrate a simple way to construct a versatile nano- immune conjugate, which allows multiple proteins or cytotoxic agents to be bound to a single albumin scaffold. Improved efficacy of the targeted drug relative to the single agents alone was demonstrated in the mouse model, which is at least in part due to altered pk of the antibody- targeted drug. Furthermore, without being bound by theory, it is believed that the versatility of the presently disclosed nano-immune conjugate that does not require a linker or target cell internalization will overcome the obstacles faced by other nanomedicines in translating results from mice to humans.
Example 9: Lyophilization of AB160
[0243] AB160 was synthesized by adding 8mg (320μ1) of bevacizumab to 20mg of ABRAXA E®. 1.66ml of 0.9% saline was then added for a final volume of 2ml for a final concentration of 4mg/ml bevacizumab and 1 0 m g/ml ABRAXANE®, and the
mixture was allowed to incubate at room temperature for 30 minutes in a 15ml polypropylene conical tube.
[0244] After the 30 minute room temperature incubation, the mixture was diluted 1 :2 in 0.9% saline to 2mg/ml and 5mg/ml bevacizumab and ABRAXA E®, respectively. These are the concentrations of the 2 drugs when prepared by the pharmacy for administration to patients.
[0245] AB160 was divided into twenty 200μ1 aliquots in 1.5 ml polypropylene eppendorfs and frozen at -80 °C.
[0246] Once frozen, the aliquots were lyophilized overnight with the Virtis 3L benchtop lyophilizer (SP Scientific, Warmister, PA) with the refrigeration on. A lyophilized preparation was generated.
[0247] The dried aliquots were stored at room temperature in the same 1.5ml
polypropylene eppendorfs. These samples were readily reconstituted in saline at room temperature for 30 minutes, followed by centrifugation for 7 minutes at 2000x g. The resulting sample was then resuspended in the appropriate buffer, as needed.
[0248] By comparison, a sample that was dried with heat and a speed vacuum was impossible to reconstitute.
Example 10: Testing of lyophilized preparations
[0249] Samples were reconstituted at different time points after lyophilization and tested for their physical properties against ABX, and freshly made AB160.
[0250] Particle size distribution was evaluated as described above.
[0251] VEGF binding was evaluated by incubation of the sample with VEGF for 2 hours at room temperature, centrifuged at 2000 x g for 7 minutes. The amount of VEGF bound to the pellet (corresponding to the nanoparticles) or remaining in the supernatant was measured with ELISA.
[0252] Paclitaxel activity was assessed by cytotoxicity against A375 cells in vitro.
[0253] Surprisingly, lyophilization did not significantly affect either the particle size, VEGF binding, or the activity of paclitaxel as shown by the ability to inhibit cancer cell proliferation. This result held for lyophilized samples stored for 1 month (FIGs. 8A-8C) or 10 months (FIGs. 8D-8F).
[0254] Further surprising is that these results were observed with nanoparticles lyophilized without the use of cryoprotectants or other agents that may adversely affect human therapeutic use.
Example 11: Efficacy of AB160 in Humans
[0255] AB160 was tested in a phase 1, first-in-man, clinical trial testing the safety of AB160 administered to patients with metastatic malignant melanoma that have failed prior therapies. The study utilizes a classical 3+3, phase 1 clinical trial design, testing 3 different doses of AB160 in the following schema:
Table 4

[0256] *Dose level 1 refers to the starting dose.
[0257] The doses were selected relevant to doses of ABRAXA E® currently used in clinical practice. AB 160 was made prior to each treatment dose. Treatments were administered as a 30 minute intravenous infusion on days 1, 8 and 15 of a 28-day treatment cycle. Treatments were continued until intolerable toxicity, tumor progression or patient refusal. Prior to every treatment cycle, patients were evaluated for toxicity; tumor evaluations were performed every other cycle (RECIST).
[0258] The study is accompanied by formal (in-patient) pharmacokinetic studies associated with dose 1 of cycles 1 and 2 of therapy.
[0259] Five patients have been administered AB 160, at 100 mg/m2 of ABX and 40 mg/m2 of BEV, of which four have been analyzed.
Table 5: Disease course in Phase I study*

information as provided in application no. PCT/US2015/054295 filed Oct 6, 2015. All patients are still living.
[0260] PFS refers to median progression free survival, i.e. the number of days of treatment before the cancer recurred. Adverse events are listed below. There was no dose limiting toxicity (DLT), i.e. the adverse events were not linked to the dose of AB 160. More detail is provided in Table 6.
Table 6: Adverse events in Phase I study


The mean PFS was 7.6 months and the median was 7.0 months. Comparison with other clinical trials
The following table shows other published clinical studies for taxane therapy for metastatic melanoma.

[0262] Spilter also examined patients who had not been previously treated, while the current study examined patients who had failed previous treatments. Ineffective prior treatment not only takes time from the expected PFS, but selects for cancer cells that are more resistant to treatment, and typically leaves a patient in poorer physical condition. Thus, the PFS for a population of patients on a "rescue" therapy (as here, with AB 160) is expected to have a lower PFS than a naive population. This can be seen in a Phase 2 clinical trial (Hersh et al., Cancer, January 2010, 1 16: 155) that examined both rescue and naive patients with
ABRAXANE® alone. For previously treated patients with ABRAXANE® alone, the PFS was 3.5 months. Hersh et dX. Ann. Oncol 2015, (epub September 26, 2015), reported a 4.8 month PFS for naive patients treated with ABX alone.
Table 9: Performance of AB160 in a limited study against published data

[0264] Thus, the ABX/BEV complex (AB160) is superior to sequential administration of ABX and BEV, or ABX alone, and achieves this superior result with a very low effective dose of BEV. The data is therefore consistent with AB160 having improved targeting of the chemotherapeutics to the tumor, and that this targeting is mediated by BEV. It is possible that the ABX nanoparticle aids in targeting the BEV to the tumor, as albumin is selectively taken up by tumors. It is also possible that the existence of the BE ABX complex shows greater stability in vivo than ABRAXANE®.
Example 12: Follow up study to investigate whether pretreatment with BEV improves targeting
6
[0265] Following the general protocol above, athymic nude mice were injected with 1 x 10 A375 human melanoma cells in the right flank and then treated with PBS, 12 mg/kg BEV, 30 mg/kg ABX, AB160, or pretreated with 1.2 mg/kg BEV and, 24hr later, AB 160. Data is
3
represented at day 7-post and day 10 -post treatment as tumor volume in mm . F 1 1A-E track tumor size over 10 days. Only mice treated with AB160 (with or without
pretreatment with BEV) showed a reduction in average tumor volume. See also FIG. 1 1 F and FIG. 1 1 G.
[0266] The day 7-post treatment data, as summarized in Figure 1 IF, show that pretreatment with BEV was associated with a statistically significant reduction in tumor volume over control or BEV alone (p<0.0001), or ABX alone (p<0.0001).
[0267] The day 10-post treatment data, as summarized in Figure 11G, again show that pretreatment with BEV was associated with a statistically significant reduction in tumor volume over control or BEV alone (p≤S0.0001), or ABX alone (p≤0.0001). Pretreatment with
BEV before AB160 was also associated with a reduction in tumor volume over AB 160 alone (p=0.02), with complete response in two mice.
[0268] In this experiment, a 12 mg/kg dose of BEV was not therapeutic. The amount of BEV added in the pretreatment group was only 1.2 mg/kg, which is 1/10 the usual dose in mice. Yet pretreatment with a subtherapeutic dose appears to show improved efficacy of the AB160 nanoparticle. This data support the idea that pretreatment with a subtherapeutic amount of BEV can clear systemic levels of VEGF, leaving a greater relative concentration at the tumor such that tumor-associated VEGF targeting by the AB160 nanoparticles is more effective.
Example 13: Alternative means of delivering nanoparticles
[0269] It is contemplated that nanoparticles of this invention can be directly delivered to the tumor. For example, nanoparticles can be delivered via intra-arterial cannula or by direct injection into the tumor. In such embodiments, it is contemplated that large nanoparticles (e.g., 580 nm or 1130 nm) can be delivered by direct injection into or proximate to a tumor.
Example 14: Antigen Binding of Lyophilized AR160
[0270] CD20 positive Daudi lymphoma cells were labeled with fluorescent tagged anti- human CD20 or isotype matched control in panel F and A, respectively, and analyzed by flow cytometry. In the other panels, the Daudi cells were pretreated with ABX, AR160, AR160L (AR160 lyophilized and resuspended into a solution suitable for injection), or Rituxan prior to CD20 labeling. Figure 12 demonstrates that CD20 binding was specifically blocked by the AR particles and Rituxan, but not ABX alone. These results suggest that the AR binds to its CD20 ligand on these cells blocking binding of the fluorescent anti-CD20.
[0271] Figure 13 is a histogram overlay of the same data presented in Figure 12.
[0272] Figure 14A and 14B depicts the particle size comparisons of ABX alone relative to AR (FIG. 14A) and AT (FIG. 14B) freshly made and lyophilized.
[0273] FIG 15 presents the results of a Daudi proliferation assay comparing the toxicity of ABX and the AR particles. The data demonstrates the lyophilized and non-lyophilized nanoparticles have essentially the same toxicity in the Daudi assay.
Example 15: Fluorescent analysis of tumor accumulation of AlexaFluor 750 labeled
nanoparticles:
[0274] Mice received intravenous (IV ) injections of equal amounts of either labeled
ABRAXA E®, labeled ABRAXA E® coated with non-specific antibodies (AB IgG), or labeled ABRAXANE® coated with Rituximab (AR160). Regions of interest (ROI) 2, 3, and 4 (FIG. 16A) track tumor accumulation based on a fluorescence threshold, ROI 1, 5, and 6 (FIG. 16 A) serve as background references. Fluorescence was determined in the ROIs 24 hours post injection. FIG 16B is a bar graph of the average fluorescence per unit of tumor area of mice in all three treatment groups were determined to provide the gross tumor delivery. FIG. 16 C is a bar graph of the average fluorescence per unit of tumor area normalized by background ROI to give proportion of drug delivered to tumor versus body. The data demonstrate that
administration of AR160 nanoparticles results in an increased fluorescence as compared to ABRAXANE® alone or ABRAXANE® coated with non-specific antibodies.
Example 16: Nanoparticles having a size of 225nm
[0275] To make a nanoparticle having a size of 225nm, the particles were prepared in accordance with Example 1 but the ratio of BEV to ABRAXANE® was 4:5, i.e., 4 parts BEV and 5 parts ABRAXANE. This ratio produced nanoparticles having a size of 225nm (AB225). The effect of AB225 was assayed in animals as set forth above. FIG. 17 depicts the survival of the mice treated with a single dose of saline, BEV, ABX, AB 160 and AB225 and with AB160 with a BEV pretreatment. At 30 days post-administration the survival of mice treated with AB225, and with AB160 with or without pretreatment with BEV far exceeds the survival of mice treated with BEV alone of ABRAXANE® alone.
Claims
1. A lyophilized nanoparticle composition comprising nanoparticles having an outer surface, wherein each of the nanoparticles comprises: a) a carrier protein; b) between about 100 to about 1000 binding agents having a
hydrophobic portion and an antigen-binding portion; and c) optionally at least one therapeutic agent; said nanoparticles being lyophilized, and wherein upon reconstitution with an aqueous solution the antigen-binding portion of said binding agents are arranged on the outside surface of the nanoparticles and are capable of binding to a target in vivo and wherein the hydrophobic portion is associated with said carrier protein and wherein the binding agent is not bevacizumab, rituximab or herceptin.
2. The lyophilized nanoparticle composition of claim 1, wherein the antigen-binding portion binds to CD38, CD52, PD-L1, Ly6E, HER3/EGFR DAF, ERBB-3 receptor, CSF-1R, STEAP1, CD3, CEA, CD40, OX40, Ang2-VEGF, or VEGF.
3. The lyophilized nanoparticle composition of claim 1 wherein the hydrophobic portion is an Fc domain.
4. The lyophilized nanoparticle composition of claim 1, wherein the antigen binding portion is an aptamer, a receptor ligand, or an Fab fragment.
5. The lyophilized nanoparticle composition of claim 1 wherein the binding agent is
obinutuzumab.
6. The lyophilized nanoparticle composition of claim 1 that is stable at about 20 °C to about 25 °C for at least 3 months.
7. The lyophilized nanoparticle composition of claim 1, wherein each of the
nanoparticles comprises between about 400 and about 800 antibodies.
8. The lyophilized nanoparticle composition of claim 5, wherein the average size of the nanoparticles is between 130 nm and 800 nm, and further wherein less than 0.01% of nanoparticles in the composition have a size greater than 800 nm.
9. The lyophilized nanoparticle composition of claim 1, wherein the average
nanoparticle size in the composition is from greater than 800 nm to about 3.5 μιη.
10. The lyophilized nanoparticle composition of claim 1 which comprises a therapeutic agent, wherein the carrier protein is albumin and the therapeutic agent is paclitaxel, said nanoparticles having an average size of approximately 160 nm.
11. The lyophilized nanoparticle composition of claim 1, wherein the at least one
therapeutic agent is located inside the nanoparticle, arranged on the outside surface of the nanoparticle, or both.
12. The lyophilized nanoparticle composition of claim 1, wherein the at least one
therapeutic agent is selected from the group consisting of abiraterone,
bendamustine, bortezomib, carboplatin, cabazitaxel, cisplatin, chlorambucil, dasatinib, docetaxel, doxorubicin, epirubicin, erlotinib, etoposide, everolimus, gefitinib, idarubicin, imatinib, hydroxyurea, imatinib, lapatinib, leuprorelin, melphalan, methotrexate, mitoxantrone, nedaplatin, nilotinib, oxaliplatin, paclitaxel, pazopanib, pemetrexed, picoplatin, romidepsin, satraplatin, sorafenib, vemurafenib, sunitinib, teniposide, triplatin, vinblastine, vinorelbine, vincristine, and cyclophosphamide.
13. The nanoparticle composition of claim 1, wherein the carrier protein is selected from the group consisting of albumin, gelatin, elastin, gliadin, legumin, zein, a soy protein, a milk protein, and a whey protein.
14. The nanoparticle composition of claim 1 , wherein the antibodies arrange into a substantially single layer of antibodies on all or part of the surface of the nanoparticle.
15. The nanoparticle composition of claim 1, wherein the nanoparticles have an
average size of approximately 160 nm and a dissociation constant between about 1 x 10"11 M and about 1 x 10"9 M.
16. A lyophilized nanoparticle composition comprising nanoparticles having an outer surface, wherein each of the nanoparticles comprises: a) a carrier protein; b) between about 100 to about 1000 antibodies having a hydrophobic portion and an antigen-binding portion; and c) optionally at least one therapeutic agent; said nanoparticles being lyophilized, and wherein upon reconstitution with an aqueous solution the antigen-binding portion of said antibodies are arranged on the outside surface of the nanoparticles and are capable of binding to CD20 in vivo and the hydrophobic portion is associated with said carrier protein and wherein the binding agent is not rituximab.
17. The lyophilized nanoparticle composition of claim 16 that is stable at about 20 °C to about 25 °C for at least 3 months.
18. The lyophilized nanoparticle composition of claim 16, wherein each of the
nanoparticles comprises between about 400 and about 800 antibodies.
19. The lyophilized nanoparticle composition of claim 17, wherein the average size of the nanoparticles is between 130 nm and 800 nm, and further wherein less than 0.01% of nanoparticles in the composition have a size greater than 800 nm.
20. The lyophilized nanoparticle composition of claim 16, wherein the average
nanoparticle size in the composition is from greater than 800 nm to about 3.5 μιη.
21. The lyophilized nanoparticle composition of claim 16, wherein the at least one
therapeutic agent is located inside the nanoparticle, arranged on the outside surface of the nanoparticle, or both.
22. The lyophilized nanoparticle composition of claim 16, wherein the at least one
therapeutic agent is selected from the group consisting of abiraterone, bendamustine, bortezomib, carboplatin, cabazitaxel, cisplatin, chlorambucil, dasatinib, docetaxel, doxorubicin, epirubicin, erlotinib, etoposide, everolimus, gefitinib, idarubicin, imatinib, hydroxyurea, imatinib, lapatinib, leuprorelin, melphalan, methotrexate, mitoxantrone, nedaplatin, nilotinib, oxaliplatin, paclitaxel, pazopanib, pemetrexed, picoplatin, romidepsin, satraplatin, sorafenib, vemurafenib, sunitinib, teniposide, triplatin, vinblastine, vinorelbine, vincristine, and cyclophosphamide.
23. The nanoparticle composition of claim 16, wherein the carrier protein is selected from the group consisting of albumin, gelatin, elastin, gliadin, legumin, zein, a soy protein, a milk protein, and a whey protein.
24. The nanoparticle composition of claim 16, wherein the antibodies arrange into a
substantially single layer of antibodies on all or part of the surface of the nanoparticle.
25. The nanoparticle composition of claim 16, wherein the nanoparticles have a
11 3
dissociation constant between about 1 x 10" M and about 1 x 10" M.
26. A lyophilized nanoparticle composition comprising nanoparticles having an outer surface, wherein each of the nanoparticles comprises: a) a carrier protein; b) between about 100 to about 1000 binding agents having a hydrophobic portion and an antigen-binding portion; and c) optionally at least one therapeutic agent; said nanoparticles being lyophilized, and wherein upon reconstitution with an aqueous solution the antigen-binding portion of said antibodies are arranged on the outside surface of the nanoparticles and are capable of binding to HER2 in vivo and the hydrophobic portion is associated with said carrier protein and wherein the binding agent is not trastuzumab.
27. The lyophilized nanoparticle composition of claim 26 that is stable at about 20 °C to about 25 °C for at least 3 months.
28. The lyophilized nanoparticle composition of claim 26, wherein each of the nanoparticles comprises between about 400 and about 800 antibodies and the antibodies arrange into a substantially single layer of antibodies on all or part of the surface of the nanoparticle.
29. The lyophilized nanoparticle composition of claim 27, wherein the average size of the nanoparticles is between 130 nm and 800 nm, and further wherein less than 0.01% of nanoparticles in the composition have a size greater than 800 nm.
30. The lyophilized nanoparticle composition of claim 26, wherein the average
nanoparticle size in the composition is from greater than 800 nm to about 3.5 μιη.
31. The lyophilized nanoparticle composition of claim 26, wherein the at least one
therapeutic agent is located inside the nanoparticle, arranged on the outside surface of the nanoparticle, or both.
32. The lyophilized nanoparticle composition of claim 26, wherein the at least one
therapeutic agent is selected from the group consisting of abiraterone, bendamustine, bortezomib, carboplatin, cabazitaxel, cisplatin, chlorambucil, dasatinib, docetaxel, doxorubicin, epirubicin, erlotinib, etoposide, everolimus, gefitinib, idarubicin, imatinib, hydroxyurea, imatinib, lapatinib, leuprorelin, melphalan, methotrexate, mitoxantrone, nedaplatin, nilotinib, oxaliplatin, paclitaxel, pazopanib, pemetrexed, picoplatin, romidepsin, satraplatin, sorafenib, vemurafenib, sunitinib, teniposide, triplatin, vinblastine, vinorelbine, vincristine, and cyclophosphamide.
33. The nanoparticle composition of claim 26, wherein the carrier protein is selected from the group consisting of albumin, gelatin, elastin, gliadin, legumin, zein, a soy protein, a milk protein, and a whey protein.
34. The nanoparticle composition of claim 26, wherein the nanoparticles have a
11 9
dissociation constant between about l x 10" M and about l x 10" M.
Priority Applications (13)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| PCT/US2016/026270 WO2017176265A1 (en) | 2016-04-06 | 2016-04-06 | Carrier-binding agent compositions and methods of making and using the same |
| EP16837869.3A EP3337823A4 (en) | 2015-08-18 | 2016-08-18 | Carrier-binding agent compositions and methods of making and using the same |
| CA2995384A CA2995384A1 (en) | 2015-08-18 | 2016-08-18 | Carrier-binding agent compositions and methods of making and using the same |
| CN201680047942.2A CN108290944B (en) | 2015-08-18 | 2016-08-18 | Carrier binder compositions and methods of making and using the same |
| CN202210483706.2A CN114796130B (en) | 2015-08-18 | 2016-08-18 | Carrier binder compositions and methods of making and using the same |
| AU2016308337A AU2016308337B2 (en) | 2015-08-18 | 2016-08-18 | Carrier-binding agent compositions and methods of making and using the same |
| JP2018506415A JP6921802B2 (en) | 2015-08-18 | 2016-08-18 | Carrier Binder Composition and Methods for Making and Using It |
| PCT/US2016/047641 WO2017031368A1 (en) | 2015-08-18 | 2016-08-18 | Carrier-binding agent compositions and methods of making and using the same |
| IL256621A IL256621A (en) | 2015-08-18 | 2017-12-27 | Carrier-binding agent compositions and methods of making and using the same |
| HK18115782.0A HK1256726A1 (en) | 2015-08-18 | 2018-12-10 | Carrier-binding agent compositions and methods of making and using the same |
| JP2021025180A JP7133050B2 (en) | 2015-08-18 | 2021-02-19 | Carrier binder composition and method of making and using same |
| JP2022066452A JP2022087255A (en) | 2015-08-18 | 2022-04-13 | Carrier-binding agent compositions and methods for making and using same |
| JP2022140647A JP2022164916A (en) | 2015-08-18 | 2022-09-05 | Carrier-binding agent compositions and methods for making and using same |
Applications Claiming Priority (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| PCT/US2016/026270 WO2017176265A1 (en) | 2016-04-06 | 2016-04-06 | Carrier-binding agent compositions and methods of making and using the same |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2017176265A1 true WO2017176265A1 (en) | 2017-10-12 |
Family
ID=55802476
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/US2016/026270 WO2017176265A1 (en) | 2015-08-18 | 2016-04-06 | Carrier-binding agent compositions and methods of making and using the same |
Country Status (1)
| Country | Link |
|---|---|
| WO (1) | WO2017176265A1 (en) |
Cited By (18)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US10213513B2 (en) | 2014-06-16 | 2019-02-26 | Mayo Foundation For Medical Education And Research | Treating myelomas |
| US10279035B2 (en) | 2012-10-01 | 2019-05-07 | Mayo Foundation For Medical Education And Research | Nanoparticle complexes of paclitaxel, trastuzumab, and albumin |
| US10300016B2 (en) | 2014-10-06 | 2019-05-28 | Mayo Foundation For Medical Education And Research | Carrier-antibody compositions and methods of making and using the same |
| US10561726B2 (en) | 2015-10-06 | 2020-02-18 | Vavotar Life Sciences LLC | Methods of treating cancer using compositions of antibodies and carrier proteins with antibody pretreatment |
| US10618969B2 (en) | 2016-04-06 | 2020-04-14 | Mayo Foundation For Medical Education And Research | Carrier-binding agent compositions and methods of making and using the same |
| US10765741B2 (en) | 2011-05-09 | 2020-09-08 | Mayo Foundation For Medical Education And Research | Methods for treating VEGF-expressing cancer using preformed nanoparticle complexes comprising albumin-bound paclitaxel and bevacizumab |
| US11040027B2 (en) | 2017-01-17 | 2021-06-22 | Heparegenix Gmbh | Protein kinase inhibitors for promoting liver regeneration or reducing or preventing hepatocyte death |
| US11160876B2 (en) | 2016-09-01 | 2021-11-02 | Mayo Foundation For Medical Education And Research | Methods and compositions for targeting t-cell cancers |
| US11241387B2 (en) | 2015-08-18 | 2022-02-08 | Mayo Foundation For Medical Education And Research | Carrier-binding agent compositions and methods of making and using the same |
| WO2022051414A1 (en) | 2020-09-02 | 2022-03-10 | Mayo Foundation For Medical Education And Research | Antibody-nanoparticle complexes and methods for making and using the same |
| US11305020B2 (en) | 2016-03-21 | 2022-04-19 | Mayo Foundation For Medical Education And Research | Methods for reducing toxicity of a chemotherapeutic drug |
| US11311631B2 (en) | 2016-09-06 | 2022-04-26 | Mayo Foundation For Medical Education And Research | Paclitaxel-albumin-binding agent compositions and methods for using and making the same |
| US11351254B2 (en) | 2016-02-12 | 2022-06-07 | Mayo Foundation For Medical Education And Research | Hematologic cancer treatments |
| US11427637B2 (en) | 2016-09-06 | 2022-08-30 | Mayo Foundation For Medical Education And Research | Methods of treating PD-L1 expressing cancer |
| US11548946B2 (en) | 2016-09-01 | 2023-01-10 | Mayo Foundation For Medical Education And Research | Carrier-PD-L1 binding agent compositions for treating cancers |
| US11571469B2 (en) | 2016-01-07 | 2023-02-07 | Mayo Foundation For Medical Education And Research | Methods of treating cancer with interferon wherein the cancer cells are HLA negative or have reduced HLA expression |
| US11590098B2 (en) | 2016-09-06 | 2023-02-28 | Mayo Foundation For Medical Education And Research | Methods of treating triple-negative breast cancer using compositions of antibodies and carrier proteins |
| US11878061B2 (en) | 2016-03-21 | 2024-01-23 | Mayo Foundation For Medical Education And Research | Methods for improving the therapeutic index for a chemotherapeutic drug |
Citations (13)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US5270163A (en) | 1990-06-11 | 1993-12-14 | University Research Corporation | Methods for identifying nucleic acid ligands |
| US5475096A (en) | 1990-06-11 | 1995-12-12 | University Research Corporation | Nucleic acid ligands |
| US5582981A (en) | 1991-08-14 | 1996-12-10 | Gilead Sciences, Inc. | Method for identifying an oligonucleotide aptamer specific for a target |
| US5840867A (en) | 1991-02-21 | 1998-11-24 | Gilead Sciences, Inc. | Aptamer analogs specific for biomolecules |
| US6011020A (en) | 1990-06-11 | 2000-01-04 | Nexstar Pharmaceuticals, Inc. | Nucleic acid ligand complexes |
| US6051698A (en) | 1997-06-06 | 2000-04-18 | Janjic; Nebojsa | Vascular endothelial growth factor (VEGF) nucleic acid ligand complexes |
| US6147204A (en) | 1990-06-11 | 2000-11-14 | Nexstar Pharmaceuticals, Inc. | Nucleic acid ligand complexes |
| US6180348B1 (en) | 1998-04-20 | 2001-01-30 | Weihua Li | Method of isolating target specific oligonucleotide ligands |
| US6699843B2 (en) | 1995-06-07 | 2004-03-02 | Gilead Sciences, Inc. | Method for treatment of tumors using nucleic acid ligands to PDGF |
| US20070166388A1 (en) * | 2005-02-18 | 2007-07-19 | Desai Neil P | Combinations and modes of administration of therapeutic agents and combination therapy |
| US20100112077A1 (en) * | 2006-11-06 | 2010-05-06 | Abraxis Bioscience, Llc | Nanoparticles of paclitaxel and albumin in combination with bevacizumab against cancer |
| WO2012154861A2 (en) | 2011-05-09 | 2012-11-15 | Mayo Foundation For Medical Education And Research | Cancer treatments |
| WO2014055415A1 (en) | 2012-10-01 | 2014-04-10 | Mayo Foundation For Medical Education And Research | Cancer treatments |
-
2016
- 2016-04-06 WO PCT/US2016/026270 patent/WO2017176265A1/en active Application Filing
Patent Citations (14)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US6011020A (en) | 1990-06-11 | 2000-01-04 | Nexstar Pharmaceuticals, Inc. | Nucleic acid ligand complexes |
| US5475096A (en) | 1990-06-11 | 1995-12-12 | University Research Corporation | Nucleic acid ligands |
| US6147204A (en) | 1990-06-11 | 2000-11-14 | Nexstar Pharmaceuticals, Inc. | Nucleic acid ligand complexes |
| US5270163A (en) | 1990-06-11 | 1993-12-14 | University Research Corporation | Methods for identifying nucleic acid ligands |
| US5840867A (en) | 1991-02-21 | 1998-11-24 | Gilead Sciences, Inc. | Aptamer analogs specific for biomolecules |
| US5582981A (en) | 1991-08-14 | 1996-12-10 | Gilead Sciences, Inc. | Method for identifying an oligonucleotide aptamer specific for a target |
| US6699843B2 (en) | 1995-06-07 | 2004-03-02 | Gilead Sciences, Inc. | Method for treatment of tumors using nucleic acid ligands to PDGF |
| US6051698A (en) | 1997-06-06 | 2000-04-18 | Janjic; Nebojsa | Vascular endothelial growth factor (VEGF) nucleic acid ligand complexes |
| US6180348B1 (en) | 1998-04-20 | 2001-01-30 | Weihua Li | Method of isolating target specific oligonucleotide ligands |
| US20070166388A1 (en) * | 2005-02-18 | 2007-07-19 | Desai Neil P | Combinations and modes of administration of therapeutic agents and combination therapy |
| US20100112077A1 (en) * | 2006-11-06 | 2010-05-06 | Abraxis Bioscience, Llc | Nanoparticles of paclitaxel and albumin in combination with bevacizumab against cancer |
| WO2012154861A2 (en) | 2011-05-09 | 2012-11-15 | Mayo Foundation For Medical Education And Research | Cancer treatments |
| WO2014055415A1 (en) | 2012-10-01 | 2014-04-10 | Mayo Foundation For Medical Education And Research | Cancer treatments |
| US20150246122A1 (en) * | 2012-10-01 | 2015-09-03 | Mayo Foundation For Medical Education And Research | Cancer treatments |
Non-Patent Citations (23)
| Title |
|---|
| "Remington's Pharmaceutical Sciences", 1990, MACK PUBLISHING COMPANY |
| "Remington's Pharmaceutical Sciences", 1995, MACK PUBLISHING CO. |
| ALLEN TM., CANCER, vol. 2, 2002, pages 750 - 763 |
| ALLEY, S.C. ET AL., BIOCONJUG CHEM, vol. 19, 2008, pages 759 - 765 |
| ARAKAWA ET AL., PHARM. RES., vol. 8, no. 3, 1991, pages 285 - 291 |
| CLELAND ET AL., CRITICAL REVIEWS IN THERAPEUTIC DRUG CARRIER SYSTEMS, vol. 10, no. 4, 1993, pages 307 - 377 |
| DEGUCHI, T. ET AL., CANCER RES, vol. 46, 1986, pages 3751 - 3755 |
| HARLOW; LANE: "Antibodies. A Laboratory Manual", 1988, COLD SPRING HARBOR LABORATORY |
| HERSH ET AL., ANN. ONCOL, 26 September 2015 (2015-09-26) |
| HERSH ET AL., CANCER, vol. 116, January 2010 (2010-01-01), pages 155 |
| HOBBS, S.K. ET AL., PROC NATL ACAD SCI U SA, vol. 95, 1998, pages 4607 - 4612 |
| HOOD ET AL.: "Immunology", 1984 |
| HUNKAPILLER; HOOD, NATURE, vol. 323, 1986, pages 15 - 16 |
| HUSTON ET AL., PROC. NATL. ACAD SCI. US.A., vol. 85, 1988, pages 5879 - 5883 |
| JAIN, R.K ET AL., NAT REV CLIN ONCOL, vol. 7, 2010, pages 653 - 664 |
| JULIEN, D.C. ET AL., AMBS, vol. 3, 2011, pages 467 - 478 |
| LANZAVECCHIA ET AL., EUR. J IMMUNOL., vol. 17, 1987, pages 105 |
| MAYO CLINIC: "Paclitaxel Albumin-Stabilized Nanoparticle Formulation and Bevacizumab in Treating Patients With Stage IV Melanoma That Cannot Be Removed by Surgery - Full Text View - ClinicalTrials.gov", 19 December 2013 (2013-12-19), pages 1 - 4, XP055238879, Retrieved from the Internet <URL:https://clinicaltrials.gov/ct2/show/NCT02020707?term=targeted+nanoparticle+therapy+for+advanced+melanoma&rank=1> [retrieved on 20160106] * |
| NILVEBRANT, J. ET AL., COMPUT STRUCT BIOTECHNOL J, vol. 6, 2013, pages E201303009 |
| PIKAL, M, BIOPHARM., vol. 3, no. 9, 1990, pages 26 - 30 |
| SCHRAMA, D. ET AL., NATURE REVIEWS. DRUG DISCOVERY, vol. 5, 2006, pages 147 - 159 |
| SCIENCE, vol. 242, 1988, pages 423 - 426 |
| YUAN, F. ET AL., CANCER RES, vol. 55, 1995, pages 3752 - 3756 |
Cited By (48)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US10765741B2 (en) | 2011-05-09 | 2020-09-08 | Mayo Foundation For Medical Education And Research | Methods for treating VEGF-expressing cancer using preformed nanoparticle complexes comprising albumin-bound paclitaxel and bevacizumab |
| US10471145B2 (en) | 2012-10-01 | 2019-11-12 | Mayo Foundation For Medical Education And Research | Methods for treating cancer using nanoparticle complexes of paclitaxel, rituximab, and albumin |
| US10441656B2 (en) | 2012-10-01 | 2019-10-15 | Mayo Foundation For Medical Education And Research | Nanoparticle complexes of paclitaxel, cetuximab, and albumin, and methods of making the same |
| US11648311B2 (en) | 2012-10-01 | 2023-05-16 | Mayo Foundation For Medical Education And Research | Cancer treatments |
| US10279035B2 (en) | 2012-10-01 | 2019-05-07 | Mayo Foundation For Medical Education And Research | Nanoparticle complexes of paclitaxel, trastuzumab, and albumin |
| US10413606B2 (en) | 2012-10-01 | 2019-09-17 | Mayo Foundation For Medical Education And Research | Methods for treating cancer with nanoparticle complexes of albumin-bound paclitaxel and anti-VEGF antibodies |
| US10376579B2 (en) | 2012-10-01 | 2019-08-13 | Mayo Foundation For Medical Education And Research | Nanoparticle complexes of albumin, paclitaxel, and anti-VEGF antibody for treatment of cancer |
| US10279036B2 (en) | 2012-10-01 | 2019-05-07 | Mayo Foundation For Medical Education And Research | Antibody-albumin nanoparticle complexes comprising albumin, bevacizumab, and paclitaxel, and methods of making and using the same |
| US10507243B2 (en) | 2012-10-01 | 2019-12-17 | Mayo Foundation For Medical Education And Research | Nanoparticle complexes of rituximab, albumin and pacltaxel |
| US10376580B2 (en) | 2012-10-01 | 2019-08-13 | Mayo Foundation For Medical Education And Research | Methods of treating cancer with antibody-albumin nanoparticle complexes comprising albumin, trastuzumab, and paclitaxel |
| US10420839B2 (en) | 2012-10-01 | 2019-09-24 | Mayo Foundation For Medical Education And Research | Methods for treating CD52-expressing cancers using compositions comprising nanoparticle complexes of paclitaxel, alemtuzumab, and albumin |
| US10406224B2 (en) | 2012-10-01 | 2019-09-10 | Mayo Foundation For Medical Education And Research | Nanoparticle complexes of paclitaxel, trastuzumab, and albumin |
| US10668151B2 (en) | 2012-10-01 | 2020-06-02 | Mayo Foundation For Medical Education And Research | Nanoparticle complexes of albumin, paclitaxel, and panitumumab for treatment of cancer |
| US10478495B2 (en) | 2012-10-01 | 2019-11-19 | Mayo Foundation For Medical Education And Research | Methods for treating cancer using nanoparticle complexes of paclitaxel, cetuximab, and albumin |
| US10493150B2 (en) | 2012-10-01 | 2019-12-03 | Mayo Foundation For Medical Education And Research | Nanoparticle complexes of paclitaxel, alemtuzumab, and albumin |
| US10213513B2 (en) | 2014-06-16 | 2019-02-26 | Mayo Foundation For Medical Education And Research | Treating myelomas |
| US11285221B2 (en) | 2014-06-16 | 2022-03-29 | Mayo Foundation For Medical Education And Research | Treating myelomas |
| US10772833B2 (en) | 2014-10-06 | 2020-09-15 | Mayo Foundation For Medical Education And Research | Albumin-CTLA-4 paclitaxel nanopartilce complex compositions and methods of making and using the same |
| US11433023B2 (en) | 2014-10-06 | 2022-09-06 | Mayo Foundation For Medical Education And Research | Albumin-PD-1 paclitaxel nanoparticle complex compositions and methods of making and using the same |
| US10610484B2 (en) | 2014-10-06 | 2020-04-07 | Mayo Foundation For Medical Education And Research | Methods of using albumin-CD20 paclitaxel nanoparticle complex compositions for treating cancer |
| US10300016B2 (en) | 2014-10-06 | 2019-05-28 | Mayo Foundation For Medical Education And Research | Carrier-antibody compositions and methods of making and using the same |
| US10624846B2 (en) | 2014-10-06 | 2020-04-21 | Mayo Foundation For Medical Education And Research | Lyophilized compositions comprising albumin-antibody paclitaxel nanoparticle complexes |
| US10596111B2 (en) | 2014-10-06 | 2020-03-24 | Mayo Foundation For Medical Education And Research | Methods of making lyophilized compositions comprising albumin-VEGF paclitaxel nanoparticle complexes |
| US10596112B2 (en) | 2014-10-06 | 2020-03-24 | Mayo Foundation For Medical Education And Research | Methods of using albumin-antibody nanoparticle complex compositions for treating cancer |
| US10391055B2 (en) | 2014-10-06 | 2019-08-27 | Mayo Foundation For Medical Education And Research | Carrier-antibody compositions and methods of making and using the same |
| US10780050B2 (en) | 2014-10-06 | 2020-09-22 | Mayo Foundation For Medical Education And Research | Lyophilized compositions comprosing albumin-EGFR paclitaxel nanoparticle complexes |
| US10780049B2 (en) | 2014-10-06 | 2020-09-22 | Mayo Foundation For Medical Education And Research | Lyophilized compositions comprising albumin-rankl or albumin-GD2 paclitaxel nanoparticle complexes |
| US10966923B2 (en) | 2014-10-06 | 2021-04-06 | Mayo Foundation For Medical Education And Research | Carrier-antibody compositions and methods of making and using the same |
| US10993912B2 (en) | 2014-10-06 | 2021-05-04 | Mayo Foundation For Medical Education And Research | Carrier-antibody compositions and methods of making and using the same |
| US10993911B2 (en) | 2014-10-06 | 2021-05-04 | Mayo Foundation For Medical Education And Research | Carrier-antibody compositions and methods of making and using the same |
| US10322084B2 (en) | 2014-10-06 | 2019-06-18 | Mayo Foundation For Medical Education And Research | Carrier-antibody compositions and methods of making and using the same |
| US11241387B2 (en) | 2015-08-18 | 2022-02-08 | Mayo Foundation For Medical Education And Research | Carrier-binding agent compositions and methods of making and using the same |
| US11660339B2 (en) | 2015-10-06 | 2023-05-30 | Mayo Foundation For Medical Education And Research | Methods of treating cancer using compositions of antibodies and carrier proteins with antibody pretreatment |
| US10561726B2 (en) | 2015-10-06 | 2020-02-18 | Vavotar Life Sciences LLC | Methods of treating cancer using compositions of antibodies and carrier proteins with antibody pretreatment |
| US11571469B2 (en) | 2016-01-07 | 2023-02-07 | Mayo Foundation For Medical Education And Research | Methods of treating cancer with interferon wherein the cancer cells are HLA negative or have reduced HLA expression |
| US11351254B2 (en) | 2016-02-12 | 2022-06-07 | Mayo Foundation For Medical Education And Research | Hematologic cancer treatments |
| US11305020B2 (en) | 2016-03-21 | 2022-04-19 | Mayo Foundation For Medical Education And Research | Methods for reducing toxicity of a chemotherapeutic drug |
| US11878061B2 (en) | 2016-03-21 | 2024-01-23 | Mayo Foundation For Medical Education And Research | Methods for improving the therapeutic index for a chemotherapeutic drug |
| US10618969B2 (en) | 2016-04-06 | 2020-04-14 | Mayo Foundation For Medical Education And Research | Carrier-binding agent compositions and methods of making and using the same |
| US11655301B2 (en) | 2016-04-06 | 2023-05-23 | Mayo Foundation For Medical Education And Research | Carrier-binding agent compositions and methods of making and using the same |
| US11160876B2 (en) | 2016-09-01 | 2021-11-02 | Mayo Foundation For Medical Education And Research | Methods and compositions for targeting t-cell cancers |
| US11548946B2 (en) | 2016-09-01 | 2023-01-10 | Mayo Foundation For Medical Education And Research | Carrier-PD-L1 binding agent compositions for treating cancers |
| US11311631B2 (en) | 2016-09-06 | 2022-04-26 | Mayo Foundation For Medical Education And Research | Paclitaxel-albumin-binding agent compositions and methods for using and making the same |
| US11427637B2 (en) | 2016-09-06 | 2022-08-30 | Mayo Foundation For Medical Education And Research | Methods of treating PD-L1 expressing cancer |
| US11590098B2 (en) | 2016-09-06 | 2023-02-28 | Mayo Foundation For Medical Education And Research | Methods of treating triple-negative breast cancer using compositions of antibodies and carrier proteins |
| US11872205B2 (en) | 2016-09-06 | 2024-01-16 | Mayo Foundation For Medical Education And Research | Methods of treating triple-negative breast cancer using compositions of antibodies and carrier proteins |
| US11040027B2 (en) | 2017-01-17 | 2021-06-22 | Heparegenix Gmbh | Protein kinase inhibitors for promoting liver regeneration or reducing or preventing hepatocyte death |
| WO2022051414A1 (en) | 2020-09-02 | 2022-03-10 | Mayo Foundation For Medical Education And Research | Antibody-nanoparticle complexes and methods for making and using the same |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| AU2020201289B2 (en) | Carrier-antibody compositions and methods of making and using the same | |
| US11655301B2 (en) | Carrier-binding agent compositions and methods of making and using the same | |
| US10596112B2 (en) | Methods of using albumin-antibody nanoparticle complex compositions for treating cancer | |
| AU2016308337B2 (en) | Carrier-binding agent compositions and methods of making and using the same | |
| US20220183980A1 (en) | Carrier-binding agent compositions and methods of making and using the same | |
| WO2017176265A1 (en) | Carrier-binding agent compositions and methods of making and using the same |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 16717752 Country of ref document: EP Kind code of ref document: A1 |
|
| 122 | Ep: pct application non-entry in european phase |
Ref document number: 16717752 Country of ref document: EP Kind code of ref document: A1 |